Purpose
Horizontal gene transfer (HGT) is the exchange of DNA between an organism and another organism that is not its offspring. It can lead to the rapid acquisition of novel functional traits in the recipient species, leaving distinctive genomic patterns behind in the process. While not all HGT events are maintained in a genome or lead to adaptive benefit, looking for patterns of HGT across a diverse array of organisms is one way we can survey for functional novelty. Many tools exist for computational discovery of HGT events from genome sequencing data, targeting different genomic patterns and with varying sensitivity, specificity, speed, and scalability. We designed the preHGT pipeline to be a flexible and rapid tool for pre-screening genomes for HGT events. Our goal was to create a pipeline to screen for putative HGT events in as many genomes as are publicly available, or that become available in the future. We wanted an approach that could successfully screen eukaryotic, bacterial, and archaeal genomes and that could screen for transfer events between closely or distantly related species.
The preHGT pipeline uses multiple existing methods for HGT screening and the elimination of false positives. It quickly produces a candidate list of genes that researchers can further investigate with more stringent HGT detection methods, different data modalities, or wet lab experimentation.
We hope this pipeline will be useful to researchers interested in exploring HGT in RefSeq or GenBank genomes.
- This pub is part of the platform effort, “Software: Useful computing at Arcadia.” Visit the platform narrative for more background and context.
- The preHGT pipeline is available in this GitHub repository.
The context
Adaptation and evolutionary innovation often occur through vertical inheritance and gradual evolutionary processes. Lateral transmission of genomic sequences via HGT is a contrasting evolutionary process that occurs between species instead of from parent to offspring. When genes are transferred, HGT can be a source of rapid functional innovation. Not all HGT events lead to adaptation — some may be neutral, detrimental, or may not be maintained by natural selection and are subsequently lost 1. Nevertheless, HGT has been the underlying mechanism for many functional adaptations 2–3.
HGT occurs across all domains of life with different frequencies and via many different mechanisms 4–6. In bacteria, HGT most frequently occurs via transduction, conjugation, or transformation. As asexual reproducers with dedicated machinery for HGT, horizontal transfer is one of the most prominent mechanisms for quickly generating genetic diversity. This can catalyze rapid evolution and adaptation to different environmental conditions 3. However, bacteria also combat HGT by degrading foreign DNA with restriction enzymes and CRISPR 7–8. Although eukaryotes can undergo HGT through transposable elements, hybridization, and viral transfer, the rate of HGT is relatively low compared to bacteria 5. This is in part due to structural barriers such as the nucleus that impede the transfer of foreign DNA into the recipient's genome. In sexually reproducing eukaryotes, the frequency of successful horizontal transfer is further reduced because foreign genomic material must reach germline cells to be transmitted from parent to offspring 9.
Surprisingly, HGT events leave behind similar signatures in recipient genomes independent of the domain of life in which the transfer event occurred. When a gene is transferred, the gene has a different evolutionary history than that of other genes in the recipient's genome. This manifests in different ways depending on how closely related the donor species is to the recipient species. The transferred gene may conserve the function of the gene in the donor genome, may carry a transfer-associated gene annotation, may be abnormally distributed in the species pangenome, or may deviate from species-specific expectations in GC content or other characteristics 10. The strength of these signals often depends on how much time has passed since the transfer event. Transferred DNA undergoes a process called amelioration, whereby the sequence accumulates mutations over time and becomes less and less distinguishable from the recipient’s genome and more and more different from the donor’s genome 10. Other evolutionary processes can further scramble the strength or clarity of a transfer event signature. For example, if many speciation events occurred since the time of the transfer event, it may be difficult to determine whether a horizontal transfer event occurred or if the incongruent evolutionary history is due to other evolutionary processes such as incomplete lineage sorting 11. If multiple transfer events of the same gene have occurred, or if there have been gene duplications and losses post-speciation, the evolutionary history of a gene may be even more difficult to disentangle. Lastly, convergent evolution and genome contamination can confound HGT discovery by genome sequence analysis as these processes can leave behind similar genomic signatures as bona fide HGT events 12–14.
Given this variation, detecting HGT in genome sequence data can be difficult, or at the very least, may require multiple strategies to find different types of transfer events. Luckily, researchers have developed many computational methods to interrogate the genomic signatures left behind in genome sequence data by HGT in different ways (Table 1). These methods fall into two general categories: parametric and phylogenetic 10.
| Tool | Category | Taxonomic scope | Event scope | Summary |
|---|---|---|---|---|
| Alien_hunter 15 | Parametric | Bacteria & archaea | Composition | Interpolated variable order motifs from compositional biases to identify and predict horizontally transferred regions in genomic sequences. |
| Alienness 16 | Phylogenetic implicit | All | Kingdom | Measures alien index and HGT score from BLASTp results on a web server. |
| APP 17 | Phylogenetic implicit | Bacteria | Pangenome | Alienness by Phyletic Pattern; Phyletic pattern of query gene distribution in closely related genomes. |
| AnGST 18 | Phylogenetic explicit | All | All | Analyzer of Gene and Species Trees; Compares gene trees to species trees and identifies discrepancies under a generalized parsimony criterion. |
| AvP 19 | Phylogenetic explicit | All | All | Alienness vs Predictor; Finds homologous sequences, produces multiple alignments, and constructs a phylogeny to analyze the topology for HGT. |
| BLAST2HGT 20 | Phylogenetic implicit | All | Kingdom | Measures alien index, donor distribution index, and bit score differences from BLASTp results. |
| DarkHorse 21 | Phylogenetic implicit | All | Kingdom, sub-kingdom | Measures lineage probability index from BLASTp results. |
| GeneMates 22 | Phylogenetic implicit | Bacteria | Pangenome | Network analysis from gene presence-absence and SNP variants. |
| GIPSy 23 | Parametric, phylogenetic implicit | Bacteria | Composition | Genomic Island Prediction Software; Predicts genomic islands using features such as abnormal GC content and presence of mobility genes. |
| HGT-DB 24 | Parametric | Bacteria & archaea | Composition | A database of potential HGT events detected using deviations in GC content and codon and amino acid usage. |
| HGT-Finder 25 | Phylogenetic implicit | All | Sub-kingdom | Measures transfer index from BLASTp results. |
| HGTector 26 | Phylogenetic implicit | All | Sub-kingdom | Measures likelihood of HGT from between self and close & distal groups from BLASTp results. |
| HGTphyloDetect 27 | Phylogenetic implicit, phylogenetic explicit | All | All | Measures alien index and out_pct from BLASTp results, followed by phylogenetic inference on initial candidates. |
| HGTree 28 | Phylogenetic explicit | Bacteria & archaea | All | A database of potential HGT events inferred using tree reconciliation. |
| Islander 29 | Parametric | Bacteria | Bacteria | Targeted identification of tDNAs. |
| IslandHunter 30 | Parametric | Bacteria | Composition | Predicts genomic islands using features such as abnormal GC content and presence of mobility genes. |
| IslandPath-DIMOB 31 | Parametric | Bacteria | Composition | Predicts genomic islands using dinucleotide composition and presence of mobility genes. |
| IslandPick 32 | Phylogenetic implicit | Bacteria | Species, Strain | Predicts genomic islands by comparing closely related genomes. |
| IslandViewer4 33 | Parametric, phylogenetic implicit | Bacteria & archaea | See other tools | Integrates IslandPick, IslandPath-DIMOB, SIGI-HMM, and Islander |
| Near HGT 34 | Phylogenetic implicit | Bacteria | Species, Strain | Measures synteny index and constant relative mutability from comparisons |
| PGAP-X 35 | Phylogenetic implicit | Bacteria | Pangenome | Pan-genome Analysis Pipeline; Pangenome gene presence absence |
| RANGER-DTL 36 | Phylogenetic explicit | All | All | Rapid ANalysis of Gene family Evolution using Reconciliation-DTL; Reconciles gene and species trees to detect duplications, transfers, and losses. |
| RecentHGT 37 | Phylogenetic implicit | Bacteria & archaea | Species, Strain | Expectation maximization algorithm on global protein sequence alignments |
| RIATA-HGT 38 | Phylogenetic explicit | All | All | Identifies incongruencies between gene trees and species trees. |
| SIB 39 | Parametric | Bacteria & archaea | Species, Strain | Sequential Information Bottleneck; Signals derived from k-mer co-occurrence to identify transferred regions |
| ShadowCaster 40 | Parametric, phylogenetic explicit | Bacteria & archaea | Composition | Uses a support vector machine on compositional features to identify candidates and then filters results by assessing ortholog similarity at increasing taxonomic distances. |
| SigHunt 41 | Parametric | Eukaryotes | Composition | Sliding window of 4-mer frequencies. |
| SIGI-HMM 42 | Parametric | Bacteria & archaea | Composition | Predicts genomic islands using a combination of codon usage bias and hidden Markov models. |
| T-REX 43 | Phylogenetic explicit | All | All | Tree-based search for Reticulate Evolution; Incongruities in phylogenetic trees |
| TF-IDF 44 | Parametric | Bacteria & archaea | Species, Strain | Term frequency-inverse document frequency to identify unusual sequence features |
Table 1. Non-exhaustive list of computational tools for HGT discovery.
Composition: Composition different from acceptor genome.
Pangenome: Any set of organisms one can reasonably build a pangenome from (clade, species, genus).
Kingdom: Cross-kingdom detection, usually by user-defined definition of ingroup and outgroup.
Sub-kingdom: Any taxonomic level lower than kingdom and higher than species or strain, usually with decreasing accuracy at higher taxonomic resolution.
Parametric methods analyze the genome of interest to identify regions that deviate from species-specific expectations in GC content, codon usage, amino acid usage, k-mer frequencies, gene annotations, or other characteristics 10. These methods are fast, but natural differences in genome uniformity can lead to over-prediction and they're often limited to recent transfer events for which amelioration of transferred DNA is limited 10. Parametric approaches can also be biased by gene length 45–46, so they may be difficult or impossible to use on genes, which vary in size, as opposed to sliding windows across the genome, which are a consistent length.
Phylogenetic methods detect inconsistencies between gene and species evolution 10. This category can be further divided into explicit and implicit methods. Explicit methods test alternative evolutionary scenarios using tree-based analysis, while implicit methods rely on implied phylogenetic relationships derived from comparative genomic approaches. Gene-by-gene explicit phylogenetic methods are the gold standard in horizontal gene transfer detection 10, 47. The most robust of these approaches works by formally reconciling gene family tree topologies (where each tip is a protein sequence belonging to a species) with the species tree topology (each tip is a species) under explicit Maximum Likelihood inference for models of gene family duplication, transfer, and loss 48–49. These methods identify candidate ancestral HGT events while accounting for the confounding impacts of gene duplication and loss on these inferences. Although powerful, these methods require that gene homology is already known and that gene family trees of these homologous sequences have already been inferred. Consequently, these methods are typically ideally suited for focused application to a set of gene families of special interest and thus are less computationally tractable to apply at scale than other HGT prediction methods.
Without a priori knowledge about the donor and recipient genomes for horizontally transferred genetic material, it becomes necessary to sample in a taxonomically broad and unbiased manner. In this respect, implicit phylogenetic methods are particularly well suited to hypothesis-free discovery of HGT events, as they scale more readily to hundreds of genomes than do explicit methods. Implicit methods rely on patterns that correlate with evolutionary history to infer HGT. For example, you can use BLAST to identify homologous genes with different taxonomic labels than the query gene, which can be analyzed to find patterns consistent with HGT 19, 25, 50–52. Similarly, you can use the pangenome — the full complement of genes shared between a set of closely related organisms — to investigate HGT by determining the presence or absence of genes across all genomes 53–54.
Across the HGT literature and tool space, including both parametric and phylogenetic methods, genome contamination is often underappreciated. Contaminant sequences in genomes can look like HGT events. This has led to rebuttals 14, 55 against high-profile papers 56–57 that claimed detection of high fractions of horizontally transferred genes, and may more generally impact the biological interpretation of HGT predictions. At least 0.54% of genomes in GenBank and 0.34% in RefSeq are contaminated 58. While some methods incorporate careful contamination checks 19, others rely on filtering heuristics 16 or omit them entirely.
The problem
We sought a scalable computational approach for predicting HGT candidate genes. We wanted the pipeline to be able to screen for HGT events across the tree of life and across taxonomic scopes (from family- to kingdom-level transfers), and to assess the likelihood that a candidate transfer event was instead the result of genome contamination.
As other projects at Arcadia are developing explicit phylogenetic methods for the inference of gene family evolution, we sought a solution that we could use upstream of this tool to produce candidate species lists for further validation, and tried to avoid using trees so as not to duplicate efforts.
Our solution
We built a pipeline that we’re calling “preHGT” to quickly find preliminary HGT candidates in genomes with gene predictions (RRID: SCR_027232). Our approach blends parametric and phylogenetic implicit methods to generate a list of candidate genes that may have been horizontally transferred (Figure 1). The preHGT pipeline uses compositional scans, pangenome inference, and BLAST-based searches. It combines information from these approaches, as well as annotation information, to highlight candidate genes that are more likely to be contamination than HGT. By implementing multiple HGT screens in one pipeline, we aimed to combine approaches that target different signatures of HGT, to provide a more comprehensive HGT screening strategy.
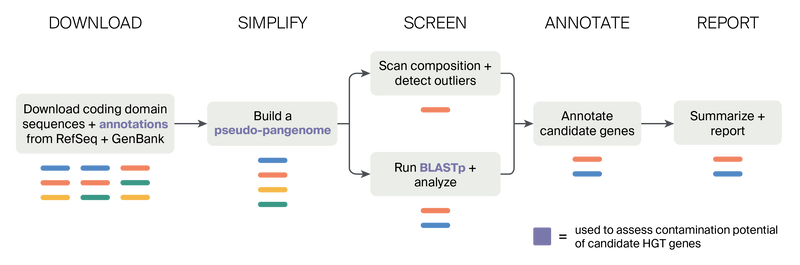
Figure 1. Conceptual overview of the HGT screening approach implemented in the preHGT pipeline.
Starting from a genus or genera, preHGT scans GenBank and RefSeq and downloads matching genomes with gene models (coding domain sequences) and annotation files. The coding domain sequences are represented by colored bars, and genes of the same identity are the same color. The pipeline uses the input genes from all genomes of the same genus to build a pseudo-pangenome. These genes are provided as input to two HGT screening methods — compositional scans and BLASTp-based approaches. These steps return HGT candidates that are then annotated to predict function. Information from each of these steps is summarized and returned in a final table.
As we were designing the pipeline, we were concerned about overall run times, especially given that BLAST searches can be computationally expensive. We implemented clustering heuristics at two key places to keep the pipeline fast. First, we clustered the genes in input genomes to reduce the number of genes we investigated for HGT potential. Given our eventual goal of running this pipeline on all publicly available genomes, we wanted to assess the potential for HGT in redundant genes only once. We did this by clustering genes in closely related genomes — those of the same genus — prior to screening for HGT. Second, we clustered the NCBI BLAST non-redundant protein database, reducing its size by over half, to increase the speed of BLAST searches 59.
One of the reasons we were particularly excited to include BLAST in our pipeline was to take advantage of a rich literature of BLAST-based HGT predictor indices (Table 1, Table 2). Many creative and insightful HGT screening methods exist, each with its own strengths. However, these methods are contained in different tools. Since BLAST is the most expensive computational step of our pipeline and none of the methods rely on a clustered BLAST database, we re-implemented them in the preHGT workflow. This consolidation allows HGT screening using a single tool and a single BLAST run (Table 2).
We implemented the pipeline as both a Snakemake 60 and a Nextflow 61 workflow, with software environments controlled by conda or Docker. The modular nature of the workflow will allow us to incorporate additional methods over time.
The preHGT pipeline does not implement any new algorithms for HGT candidate screening. However, the pipeline contributes to this space by:
- Combining multiple existing HGT screening algorithms in one pipeline.
- Using pangenome inference on eukaryotic genomes to inform a gene’s contamination potential and phyletic distribution, and to reduce compute required to run the pipeline.
- Reducing the BLAST database size by clustering similar proteins, thereby reducing compute required to run the pipeline and diversifying taxonomic lineages represented in top hits.
- Providing multiple information sources to help assess an HGT candidate’s contamination potential.
The resource
The preHGT Snakemake and Nextflow workflows are available at this GitHub repository (DOI: 10.5281/zenodo.8169268, RRID: SCR_027232).
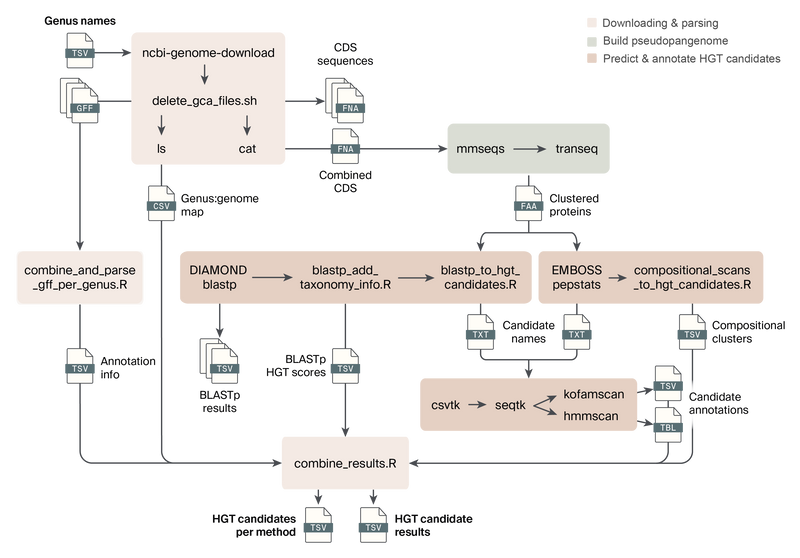
Figure 2. Overview of the preHGT pipeline steps, inputs, and outputs.
Users provide a genus or genera of interest in a TSV file as input to the pipeline. The workflow then downloads and parses the available genomes for those genera, builds a pseudo-pangenome, and predicts and annotates horizontally transferred gene candidates.
Below we provide an overview of each step in the preHGT pipeline (Figure 2).
-
Retrieving gene sequences and annotation files. The pipeline begins with the user providing a genus or genera of interest in a TSV file. The pipeline then scans GenBank 62 and RefSeq 63 for matching genomes and downloads gene models and genome annotation files using ncbi-genome-download. When a genome is available in both GenBank and RefSeq, only the RefSeq version is retained.
-
Building a pseudo-pangenome. For each genus, the pipeline then combines genes into a pseudo-pangenome by clustering the nucleotide sequences at 90% length and identity using
mmseqs easy-cluster64. For each cluster, MMSeq2 selects a single representative sequence by retaining the sequence with the most alignments. The clustered nucleotide sequences are then translated into amino acid sequences using EMBOSStranseq65. We refer to this as a pseudo-pangenome, and not a pangenome, because we empirically cluster genes based on sequence similarity and not by constructing orthologous groups or by considering the evolutionary history of each sequence 66–67. We recognize that while this may collapse functionally different paralogs, it is unlikely to obscure patterns of HGT from distant donor genomes; paralogous genes share a common ancestor, so while they may serve different purposes for the organism, at >90% identity only one copy of the gene needs to be screened for HGT potential. Using a pseudo-pangenome is useful in two ways for the pipeline. First, it reduces the number of genes that are investigated which reduces run times. Second, it provides metadata about the gene. Singletons are more likely to be contaminants, and genes that are only present in a subset of genomes may have interesting evolutionary histories (e.g., gene loss). -
Screening for HGT candidates. Using the genes in the pseudo-pangenome, the preHGT pipeline then uses two approaches to screen for HGT candidates.
-
Compositional scan. The first approach uses relative amino acid usage to detect proteins with outlying composition. It measures relative amino acid usage using the EMBOSS
pepstatsfunction 65, produces a distance matrix with the base R functiondist(), and hierarchically clusters the distance matrix with fastcluster’shclust68. It detects outliers by cutting the resultant tree withheight/1.5and retaining any cluster that contains fewer than 0.1% of the pseudo-pangenome size. Relative amino acid usage is the frequency that each amino acid is used in each gene, normalized by the total number of amino acids in that gene. For example, if alanine is used 27 times in a protein that is 100 amino acids long, the relative usage would be 27%. Relative amino acid usage is generally conserved across a genome and reflects an organism's environment 69. We tried many compositional metrics such as tetranucleotide frequency, GC content, and codon usage. However, we found that outlying proteins were driven by abnormal length for all metrics other than relative amino acid usage. Given that this is a reference-free approach, genes returned by this screening method do not have accompanying donor species predictions, which makes interpretation more challenging. Aberrant relative amino acid usage can also arise from mechanisms other than HGT and this method does not distinguish between potential sources. -
BLASTp scan. The second approach uses BLASTp to identify homologous proteins. All genes in the pseudo-pangenome are BLASTed against a clustered version of NCBI’s nr database (90% length, 90% identity) 59 using DIAMOND
blastp70. The pipeline then adds lineage information to the BLASTp search using dplyr, dbplyr, and RSQLite 71. It scans these results for signatures of transfer events using multiple, previously published algorithms (Table 2) 19, 25, 50–52. One modification we made throughout is using length-corrected bit scores output by DIAMONDblastpinstead of raw bit scores. Bit scores are sensitive to gene length, so using corrected bit scores reduces biases associated with gene length in HGT screening 72. The choice of database will dramatically impact the results produced by this screen. We chose to use a clustered version of the NCBI nr database 59 both to make the BLASTp step faster and to ensure the results contain a variety of taxonomic lineages in cases where many near and distant homologs exist. Using this database, combined with our methods of choice (Table 2), the preHGT pipeline screens for HGT events that occur in seven domains of NCBI’s taxonomy: bacteria, archaea, fungi, plants, metazoa, other eukaryotes, and viruses (“kingdom” taxonomic resolution). It will also screen for HGT events between lineages that are in the same domain as the query genus but are different up to the family level from that genus (“sub-kingdom” taxonomic resolution).Index Tool Taxonomic resolution Data used Calculated by Aggregate hit support+
19AvP Kingdom All bit scores Subtracting the sum of normalized bit scores in the donor group from the sum of normalized bit scores in the acceptor group. Alien index
50NA Kingdom Minimum E-value Subtracting the transformed E-value of the best donor hit from the transformed E-value of the best non-self acceptor hit. HGT score
51NA Kingdom Maximum bit score Subtracting the best non-self acceptor hit bit score from the best donor hit bit score and normalizing this value. Donor distribution index
52NA Kingdom Number of hits per kingdom Measuring the dispersion query homologs across groups by determining the number of hits per kingdom against the total number of possible kingdoms. Gini coefficient NA Kingdom Number of hits per kingdom Measuring inequality among values of a distribution, where values are the number of BLAST hits observed for each kingdom. Entropy NA Kingdom Number of hits per kingdom Measuring disorder among values, where values are the number of BLAST hits observed for each kingdom. Transfer index
25HGT-Finder Kingdom, Sub-kingdom All bit scores Considering taxonomic distances between query and hit, bit score ratios, and rank and total number of BLAST hits. Table 2. Algorithms that parse BLASTp results to predict HGT candidates.
*NA: Not applicable.
+Aggregate hit support is calculated by subtracting the sum of all normalized BLAST bit scores for all hits in an in-group from an out-group. We use a different normalization equation than the original method, which leads to different results.
-
-
Annotation. We then annotate the HGT candidates. For each candidate HGT amino acid sequence, we use two different approaches for ortholog annotation. First, the pipeline uses KofamScan for KEGG ortholog annotation 73. Next, the pipeline uses HMMER3
hmmscanto assign annotations to HGT candidates.hmmscancompares each HGT candidate sequence against hidden Markov models (HMMs) of proteins in a database. We built a custom HMM database to target specific annotations of interest. The HMM database currently contains Virus Orthologous Groups from VOGDB and biosynthetic genes and can be extended in the future to meet user annotation interests. -
Reporting. The last step combines all information that the pipeline has produced and outputs the results in a TSV file. The results include the GenBank protein identifier for the HGT candidate, BLAST and relative amino acid usage scores, pangenome information, gene and ortholog annotations, and contextualizing information about the gene such as position in the contiguous sequence.
Types of HGT events that the pipeline screens for
While we tried to create a fast and generalized pipeline, preHGT is better at detecting some patterns of HGT than others. The preHGT pipeline screens for HGT events where the donor and recipient differ in taxonomy at the family level or above. It is most likely more accurate when the transfer events occur between more distantly related organisms and where the recipient gene retains homology to the gene in the donor genome. We anticipate the primary use of this approach will be to identify candidate transfer events and donor and recipient groups to which more granular approaches can be applied to better disentangle the evolutionary history of the gene.
The parametric approach we implemented screens for genes with outlying relative amino acid usage compared to the rest of the genes in the genome. This requires that the donor and acceptor species differ in amino acid composition, and that these differences persist in the transferred genes, a scenario that is most typical of recent transfer events among evolutionarily divergent species.
The BLAST-based implicit phylogenetic approaches we implemented screen for genes that exhibit a greater degree of sequence similarity among designated taxonomic outgroups than within ingroups. In the original tools and papers in which these algorithms were generated, the authors implemented or validated their approaches at specific taxonomic levels that the preHGT pipeline adheres to (Table 2). Some are designed to screen for cross-kingdom transfer events, while others can screen for sub-kingdom-level events. However, because the chance of spurious inference of homology increases among more closely related species, results should be more carefully scrutinized at lower taxonomic levels (e.g., order, family). Homology detection also becomes increasingly difficult at larger taxonomic distances, so the pipeline may miss highly diverged homologs.
Additional considerations and caveats
How we deal with contamination and other sources of false positives
HGT screens often return many false positives 56–57. We used contextualizing information about HGT candidates to reduce the number of false positives reported by the pipeline.
Contamination is the biggest source of false positives in BLAST-based HGT screening algorithms. Many genomes in GenBank and RefSeq are contaminated 58. Contamination arises from impure sampling, contaminated reagents, lab cross-contamination, sequencing artifacts, or reference database errors 74. To combat the presence of contamination, we incorporated multiple corroborating lines of evidence to assess whether contamination is more likely than HGT. First, we determine the length of the contiguous sequence within which the candidate gene is found. Short contiguous sequences are more likely to be contaminants 58, 75. Next, we determine how many genes are in the candidate gene’s cluster from our pseudo-pangenome approach. Depending on the contamination source, it is unlikely that the same contamination will occur in multiple genomes 76. Therefore, if a homolog is present in multiple genomes, it is less likely to be a contaminant. Lastly, for BLAST-based results, we assess the percent identity between the donor and acceptor genes. Amelioration deteriorates sequence identity after a transfer event 10, so the more similar two genes are, the more likely similarity is driven by contamination. Many methods use a cutoff of 70%–80% identity for contamination 16, 77, but we instead weigh this against other corroborating information.
In the future, we hope to further contextualize contamination potential against the general contamination score for the acceptor genome. The more contamination a genome contains, the more likely a candidate is to be a contaminant itself.
BLAST-based methods may also generate false positives arising from alignment errors or alignment due to sequence similarity that does not arise from shared ancestry, such as from convergent evolution or random chance. Alignment errors from short or low-complexity sequences or from short, highly conserved domains may give the appearance of a horizontal transfer event. To protect against this, we filter corrected bit scores to those greater than 100, or, to rescue true homologs that are very divergent, with a query coverage of greater than 70%. We also provide gene annotations from multiple annotation sources to highlight hits that might be ultra-conserved, such as those from ribosomal proteins. Over time, we hope to curate a list of genes that the preHGT pipeline frequently detects as false positives and to develop a strategy to filter them out.
Verifying bona fide HGT requires work beyond preHGT
The preHGT pipeline provides a list of candidate HGT events. These candidates need to be carefully scrutinized to determine whether they are biologically interesting and whether they are more or less likely to be false positives. We built preHGT as a generalized precursor to more in-depth HGT analysis (Figure 3). We envision that preHGT can inform genome selection for comprehensive explicit phylogenetic inference, which can help disentangle alternate evolutionary trajectories, or highlight when not enough information is available to support HGT inference.
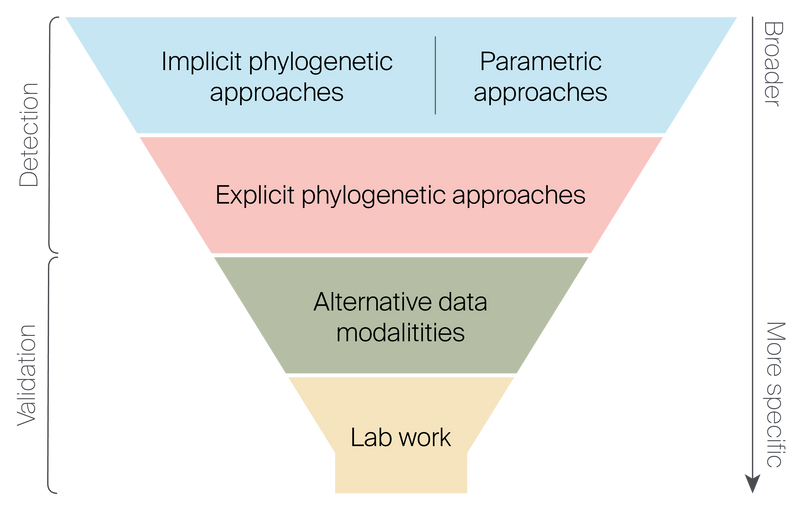
Figure 3. Funnel of methods for HGT screening and validation.
Implicit phylogenetic and parametric approaches are fast, generalized methods for screening for HGT in genes or genomes, but these methods are prone to false positives. Explicit phylogenetic methods can help eliminate some false positives or determine when there is not enough evidence to support HGT candidacy.
After these computational approaches, validation requires additional methods. Alternative data modalities like transcriptome sequencing or laboratory experiments like FISH or PCR can provide additional evidence in support of HGT. While powerful, these methods require curated information about the donor and acceptor genomes and the candidate genes and thus can usually only be used after initial exploration.
After phylogenetic analysis, more analysis is still required to reject the null hypothesis that no transfer event occurred. The appropriate experiments for this will depend on the HGT candidate event itself. For example, if a bacterial gene has been transferred into a eukaryotic genome, it may be appropriate to interrogate the candidate gene for the presence of introns, or if transcriptome information is available, for the presence of transcription- and eukaryotic-specific RNA modifications such as 5′ caps or Kozak sequences. In the lab, PCR, FISH, or Southern blots may confirm the presence of the sequence in the genome of interest, while Western blot or mass spectrometry can confirm that the gene is transcribed and translated into a protein.
Limitations of the preHGT pipeline
Given our approach, we have identified multiple shortcomings. The most conspicuous limitation is our focus on genes. The preHGT pipeline can only scan genomes with gene models. We elected not to implement genome annotation as an early step in the pipeline given that annotation procedures differ for eukaryotic versus bacterial and archaeal genomes, and that eukaryotic genome annotation remains a challenging problem from the genome alone 78–79. This limits the preHGT pipeline to those genomes with gene models (approximately 21%) and creates blind spots for HGT detection across the tree of life. Of 56 eukaryotic phyla with genomes, only 45 have at least one genome with gene models. Similarly, by treating genes in their entirety as the unit that is horizontally transferred, we are unlikely to detect genes for which only a nested region of the coding sequence was horizontally transferred.
There are also limitations born out of our decision to use composition or BLAST-based HGT screening methods. First, these methods require that the gene has not ameliorated to the composition of the acceptor genome or that it maintains detectable homology to the donor genome. This may limit our detection of ancient HGT events. Second, these methods will be less sensitive to HGT events that occur between closely related organisms. Third, since BLAST-based approaches rely on taxonomies, there are risks since taxonomies may be wrong and since they do not account for branch lengths in the relatedness of species. Lastly, false positives may arise from alignment between short or low-complexity sequences or from natural sequence similarity such as what might arise from convergent evolution or from highly conserved gene sequences. To combat both cases, we have implemented filtering criteria to help eliminate these issues.
Lastly, we did not integrate an explicit phylogenetic approach to better resolve the evolutionary histories of HGT candidates. We elected to forgo this step because another team at Arcadia is developing a tree-based workflow. We are currently experimenting with how to facilitate handoff between the two tools to rapidly enable this next step in validation.
Additional methods
We used ChatGPT to add comments to our code and suggest wording ideas. We also used ChatGPT to add comments to external code to help us better understand how it worked when trying to implement some existing tools in another language.
Key takeaways
- preHGT is a scalable pipeline that screens for potential HGT events in genomes with gene models across the tree of life and taxonomic scales.
- The pipeline leverages compositional and BLASTp scans, pangenome inference, annotation, and reporting techniques to provide comprehensive results.
- Multiple checks and filters defend against false positives, including contamination detection and sequence alignment artifact filtering.
- The pipeline is implemented in both Snakemake and Nextflow. Its modular design means it’s easily extensible to incorporate more methods in the future.
- preHGT aims to identify HGT events that users further investigate with other approaches such as tree-based ones.
Next steps
Our follow-up plans include:
- Eukaryotic HGT prediction: We plan to run the pipeline on all eukaryotic genomes in GenBank and RefSeq that have gene models and to make the results available.
- Building a user interface for results exploration: We plan to build a simple user interface to explore results produced by the pipeline. Exploration modes will allow users to dive into gene transfer events by donor or acceptor taxonomy, predicted functions of genes involved, or by strength of result, and to visualize the results in their genomic context.
- Adapting the pipeline to take transcriptome assemblies as input: We plan to extend the pipeline to run on assembled transcriptomes by incorporating upstream gene prediction rules. We will then run the pipeline on the transcriptomes in the NCBI Transcriptome Shotgun Assembly database and make the results publicly available.
- Integrating new algorithms for HGT screening: Other algorithms exist for the interpretation of BLASTp results. We plan to integrate those from other tools into this pipeline in the future.
We welcome feedback on the user experience, the results we include, or additional algorithms or metrics that would be helpful to incorporate.
Be the first to comment on this publication.