Purpose
The seqqc pipeline rapidly assesses the quality of new sequencing data. Our goal is to make sure that data is free of common quality issues without formal analysis so that we can rapidly deposit new sequencing results to make them available to others. The seqqc pipeline produces an interpretable HTML report that we’ve designed to help identify technical artifacts (e.g., sequence duplication rate), process-oriented issues (e.g., mislabeled samples), and contamination in long- and short-read sequencing data. We hope that others will find it useful to integrate seqqc into their own sequencing workflows to improve data quality and allow for quicker deposition.
- This pub is part of the project, “Useful computing at Arcadia.” Visit the project narrative for more background and context.
- The seqqc Nextflow workflow is available at this GitHub repository.
- The workflow to build the seqqc contamination database is available at this GitHub repository.
- The contamination database is available on OSF.
- We’ve included sample seqqc reports for different sequencing data types for reference (Illumina, PacBio HiFi, PacBio IsoSeq, and Nanopore).
Why it matters
We want to release our data for others to use as rapidly as possible, while making sure that the data we upload into public repositories is high-quality. We also want to catch any potential errors as early as possible to either inform our data analysis strategy or highlight additional experiments that need to be performed.
The problem
In a traditional academic setting, sequencing data is released at the time of preprint or publication. This means that the data has undergone thorough quality control during normal data analysis cycles, catching any issues that may need to be addressed — mislabeled samples, index hopping, etc. While these checks are important, the typical order of events creates a bottleneck that greatly delays the usability of these data for others in the science community. At Arcadia, we want to decouple these events by rapidly releasing new sequencing data to International Nucleotide Sequence Database Collaboration (INSDC) databases to make it fully accessible, as soon as possible. This means that data release will take place prior to formal analysis, and may have undiscovered quality issues.
Our solution
We designed a Nextflow pipeline, seqqc, to rapidly assess the quality of short- or long-read sequencing data (Figure 1). The pipeline reports technical sequencing issues via FastQC 1, sample similarity with sourmash compare 2, and potential contamination with sourmash gather 3. Each quality control module is summarized as a visualization via MultiQC HTML reporting 4. The final report contains explanations on how to interpret the results and how interpretations may change with data type. This pipeline has allowed us to do faster quality control of our sequencing data and to flip the typical order — analysis before release — to release before analysis.
The resource
The seqqc Nextflow workflow is available at this GitHub repository (DOI: 10.5281/zenodo.7650901). The workflow to build the seqqc contamination database is available at this GitHub repository (DOI: 10.5281/zenodo.7594935). The contamination database is available on OSF (DOI: 10.17605/OSF.IO/SNDZ5).
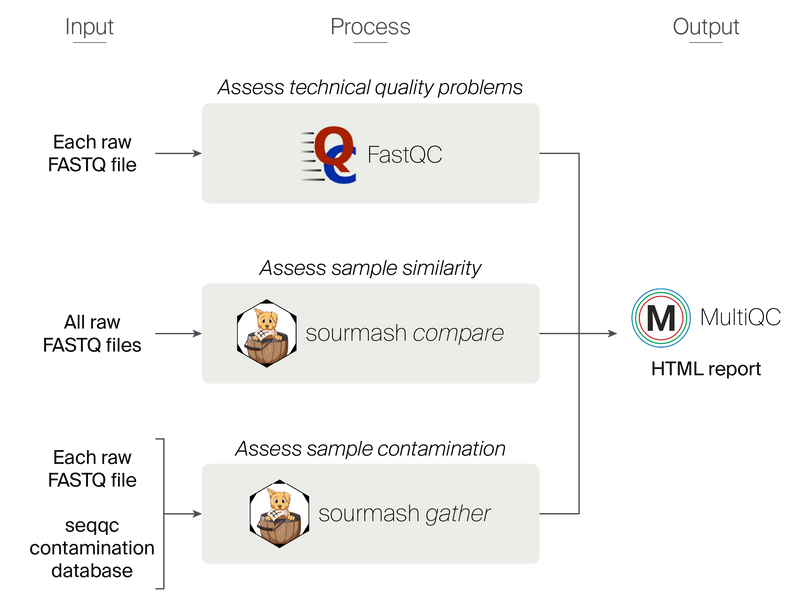
Figure 1. Overview of the seqqc workflow.
Using paths to raw FASTQ files that are documented in a CSV file, the seqqc pipeline applies three processes to assess sequence quality. A MultiQC HTML report summarizes the outputs of these processes.
An overview of the seqqc pipeline
The seqqc pipeline ingests a sample sheet that includes the sample name and the local path, URL, or URI for the raw sequencing reads in FASTQ format. FASTQ files can be short or long reads. Users can enter short reads as single or paired-end, and long reads as single-end.
The first step in the pipeline runs FastQC on each FASTQ file to assess technical artifacts that may arise during sequencing. FastQC is a field-standard quality control tool that estimates several metrics like sequencing depth, quality scores, read length, sequence duplication levels, and adapter content 1.
The second step in the pipeline runs sourmash compare on each sample to assess sample similarity 2. This module can highlight mislabeled samples or identify outlying replicate samples. sourmash compare estimates sample similarity using angular similarity. Angular similarity takes both shared sequence content and sequence abundance information into account. sourmash compare operates on FracMinHash sketches, so it uses about 1/1000th of all distinct k-mers to estimate similarity (scaled = 1000) 3. This approach preserves the accuracy of sample similarity estimates but decreases the resources required to generate the estimate 3. Sequence similarity scores range from 0 to 1 and samples that are more similar will have a higher value. Replicates or biologically similar samples should have the highest similarity scores.
The third step in the pipeline runs sourmash gather on each sample to assess potential contamination 3. sourmash gather selects the best reference genomes containing the sequences in a query by finding the smallest set of non-overlapping matches in a database 3. Contamination in sequencing data can come from many different sources. We targeted five types that are described below.
- Contamination from barcode/index hopping during Illumina sequencing. Index hopping occurs most frequently for low-biomass samples 5. To catch this type of contamination, we screen for sequences from model organisms that scientists sequence frequently (e.g., mice and E. coli). Any given flow cell will likely contain these samples at some point, making them common and easily identifiable contaminants.
- Contamination from humans handling the sample 6. This could be human sequence or sequence from microbes that live on human skin or in the mouth. We included human DNA and common human skin/oral microbiome species in the contamination database to catch this type of contamination.
- Kit contamination. Kits and reagents have their own microbiome and DNA extracted from these organisms can sneak into the sample 7. We added the most common kit contaminant organisms to the database as described in 8. Most organisms are described at the genus level, so we included all genomes below a given genus that are present in the Genome Taxonomy Database (version rs207) 9.
- Contamination caused by accidentally extracting DNA or RNA from an organism in the lab other than your own sample 10. The contamination detection database contains select species that Arcadians commonly use to try to catch this type of contamination. If users would like to include additional organisms, they can rebuild the contamination database following the included instructions for adding additional genomes. Additional genomes are specified in a pair of CSV files that record the GenBank or RefSeq genome accessions.
- Spike-in contamination 11. Illumina spikes phiX into many of its sequencing runs. We included the phiX spike in sequencing in our contamination detection database. PhiX contamination is normal and non-problematic but should be removed prior to analysis.
In total, our contamination database contains 4,515 organisms (Figure 2). For Eukaryotic genomes, we chose to build the database from GenBank- or RefSeq-predicted CDS from genomic files to remove repetitive sequences that could return false positives.
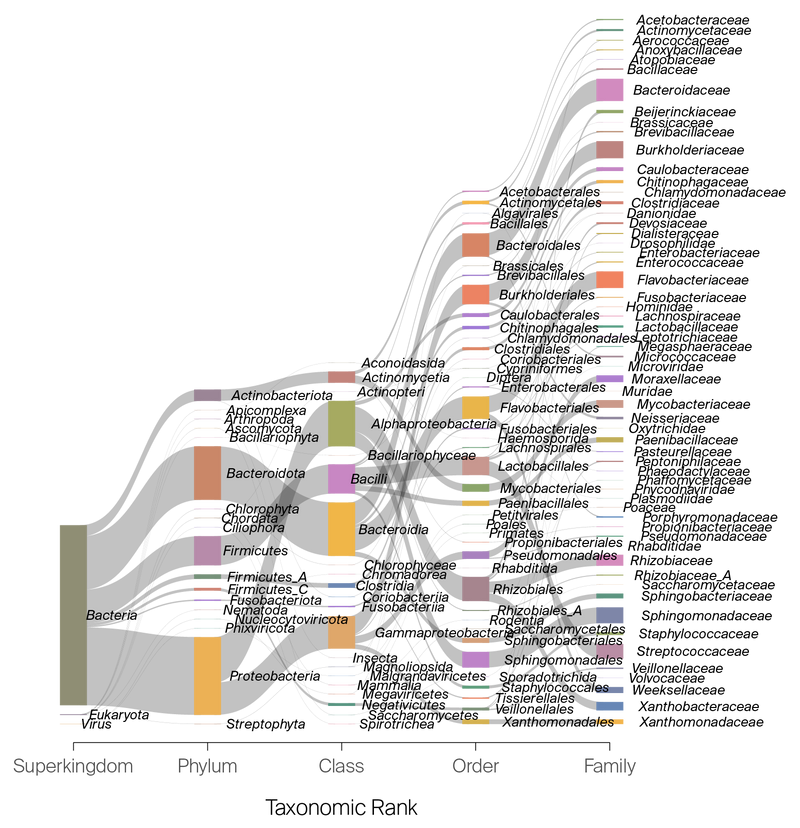
Figure 2. Sankey diagram outlining the family-level membership in the seqqc contamination database.
The database contains 4,515 species.
The last step in the pipeline summarizes the results using MultiQC 4. MultiQC parses log files from many bioinformatics tools and creates standard-format text files and interactive visualizations presented as an HTML file 4. We added comments to each section of the HTML file to describe the quality control module and how to interpret the results for a given data type — for example, it would not be alarming for a metagenome to fail the GC content module of FastQC, as we expect metagenomes to be composed of many genomes with different GC content. In contrast, this would be problematic for a single genome.
Above, we provide a preview of a MultiQC report output by seqqc for Illumina data. Browse sample reports for different types of sequencing data (Illumina, PacBio HiFi, PacBio IsoSeq, and Nanopore):
Implementation
The seqqc pipeline is a Nextflow pipeline 12. We started the pipeline using nf-core tools nf-core create 13. Our goal was to increase the interpretability of our code and reduce long-term technical debt by plugging into a vibrant open-source community 14. However, we elected not to contribute the pipeline back to nf-core and observed some tension in using the nf-core tooling downstream of this decision. We expect these issues to resolve over time, as we have observed others encountering the same barriers.
We chose a modular design to make it easy to update a given module or to add new ones as needed.
We used nf-core modules for each of our processes 13. For software tools that did not have modules in nf-core (e.g., sourmash compare), we wrote these modules and contributed them back to the nf-core/modules GitHub repository.
nf-core modules also take advantage of BioContainers, a project that partners with Bioconda to build Docker and singularity containers from conda-forge and Bioconda packages 15–16. This means the user can choose to run the seqqc pipeline with conda, Docker, or Singularity.
Similarly, we wrote MultiQC modules for sourmash compare and sourmash gather and contributed these back to the MultiQC tool.
Deployment
One reason we chose to write the seqqc pipeline in Nextflow was to access the Nextflow Tower infrastructure 14. Tower is a web application that helps manage configuration and deployment of Nextflow pipelines on various compute infrastructures. At Arcadia, we use Amazon Web Services (AWS) for our cloud computing needs, and Tower provides a seamless interface to AWS EC2 spot instances via AWS Batch, which minimizes compute costs.
We further streamlined internal execution of seqqc by implementing a cron job that periodically polls an internal AWS S3 bucket for new data, launches the pipeline on the new data, and sends an email to the uploader upon pipeline completion with the MultiQC HTML report as an attachment.
While we have engineered this architecture for Arcadians and provided documentation on the setup for other individuals or organizations who want to establish a similar setup, the seqqc pipeline itself is still fully executable locally via the command line and extensible to work with other compute infrastructure via Nextflow executors.
Key takeaways
We built a Nextflow pipeline, seqqc, that performs quality control to quickly catch problems in sequencing data to increase the pace of data release. The product of the pipeline is a set of interactive visualizations and documentation for their interpretation rendered by the popular tool MultiQC. The pipeline is built on widely-used open-source architecture (Nextflow, nf-core tools pipeline, nf-core modules, and BioContainers) so researchers can reuse the modular components and swap out or add new functionality as necessary.
The seqqc Nextflow workflow is available at this GitHub repository (DOI: 10.5281/zenodo.7650901). The workflow to build the seqqc contamination database is available at this GitHub repository (DOI: 10.5281/zenodo.7594935). The contamination database is available on OSF (DOI: 10.17605/OSF.IO/SNDZ5).
Next steps
As we use our seqqc resource on newly sequenced libraries, we expect to improve the documentation for quality control module interpretation. We welcome feedback from users who try out the pipeline.