Purpose
Our overall approach at Arcadia is to use an evolutionary lens to source novel solutions to human disease. To this end, we’ve developed a structural mimicry detection pipeline to identify cases where parasites use protein structural mimics to manipulate their human hosts’ biology, including their anti-parasite immune response. We’re starting our pipeline development using viral proteins, because viruses (especially large, double-stranded DNA viruses like herpesviruses and poxviruses 1) are well known to use mimicry to modulate host immunity 2.
We benchmarked the first version of our pipeline using well-characterized viral proteins known to mimic 11 different host proteins. For each host protein, the pipeline recovered at least one known mimic, demonstrating its ability to identify host targets of viral mimicry. While we’ve decided not to move forward with this line of research at Arcadia, this pipeline is ready for deployment by anyone who wants to identify novel parasite mimics and human targets of mimicry.
- This pub is part of the project, “Ticks as treasure troves: Molecular discovery in new organisms.” Visit the project narrative for more background and context.
- Data from this pub, including our Foldseek search results and the selected potential mimicry events, is available on Zenodo.
- The viral protein query structures we used in this work and code for processing the Foldseek search results, running Gaussian mixture models, and creating the figures for the pub are available in this GitHub repository.
We’ve put this effort on ice! 🧊
#StrategicMisalignment
We’ve decided not to pursue this project because it doesn’t play to the unique strengths of our platform. We’re sharing it in the hope that it will enable future research in this area outside of Arcadia.
Learn more about the Icebox and the different reasons we ice projects.
Background
Understanding the strategies that parasites use to manipulate their host's immune system can lead to new approaches for treating autoimmune and inflammatory diseases. Ideally, we’d follow nature’s lead and compare the targets of a wide range of parasite effectors to find common human targets amenable to drug intervention. However, parasite effectors haven’t been comprehensively characterized in the lab, and it's difficult to computationally predict the target or precise function of parasite proteins (you can see one of our attempts to do so here 3).
We hypothesized that mimicry can provide us with a shortcut to target prediction. When a parasite effector protein mimics the structure of a specific human protein, we can hypothesize that the parasite is acting on the same pathway, or may have some of the same binding partners or substrates as its human counterpart (Figure 1).
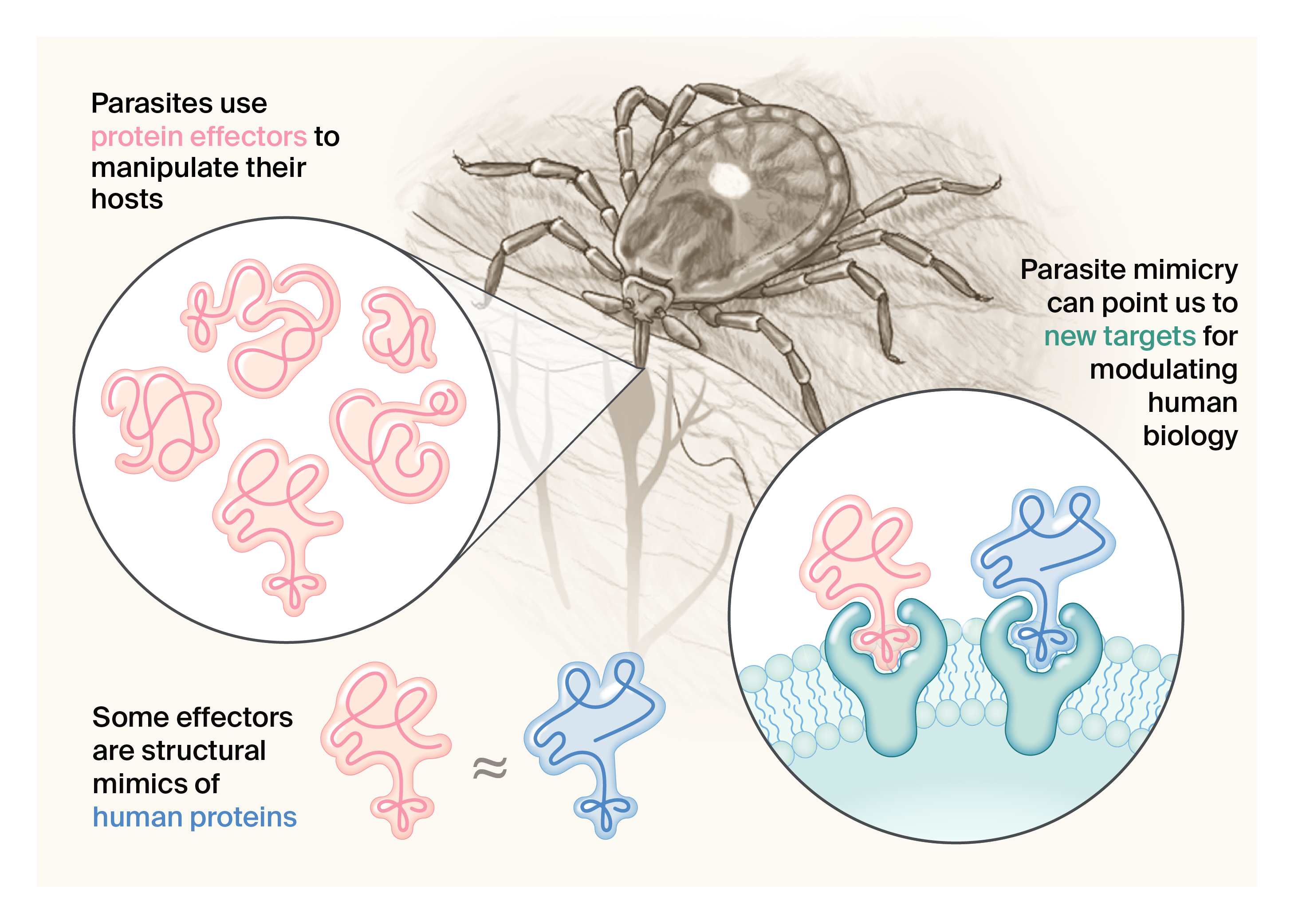
Figure 1. Ticks and other parasites like viruses use mimics to hijack host pathways and modulate host biology.
We became especially interested in mimicry after recently finding evidence that ticks may use immune-related protein mimics to manipulate their hosts (see identification of an IL-17 mimic here 3, and an SAA mimic here 4). While mimicry is thoroughly documented in viruses, it’s not well studied in ticks, making this an interesting parallel between two very different types of parasites. We decided to try using mimicry to identify commonly targeted host proteins across a wide range of parasitic species.
Where do mimics come from?
Most viral structural mimics arise from horizontal gene transfer from hosts 5, although some arise from convergent evolution 6. The origins of putative mimics from ticks are unknown.
Our first step was to build a protein structural mimicry detection pipeline using viral proteins to benchmark its performance. We decided to use viral mimics to optimize our pipeline because, unlike tick mimics, there’s a wealth of work studying viral mimics and their activities that we can use to evaluate our approach (see Table 1). We’re focused on detecting structural mimicry because shared structure often points to related function, even when the underlying sequences are different 7.
What do viral mimics do?
Mimics can have similar functions to their host counterpart (see BHRF1, a Bcl-2 mimic with anti-apoptotic activity similar to human Bcl-2 8), or they can have new, antagonistic functions (see VACWR034, an interferon-resistance protein that inhibits host PKR through mimicry of eIF2α 9). In both cases, the mimics have some shared binding partners or substrates with the host protein, but the ultimate functional outcome is different. We’re interested in mimics that act similarly as well as mimics that are antagonistic, because in both cases, they point us to important host biology.
We benchmarked the performance of our pipeline using viral proteins that fall into three different categories:
- Viral proteins that are known to mimic a specific human protein, with clear supporting experimental evidence. We call these “well-characterized mimics.”
- Viral proteins that have been described as mimics due to structural similarity to a human protein or class of proteins, but which lack experimental evidence to implicate them as mimics of one specific human protein. We call these “incompletely characterized mimics.”
- Viral proteins that we do not expect to be mimics, but that we expect to have at least partial structural similarity due to a shared function in humans and viruses. We call these “viral proteins with common domains.”
We included these three categories of viral proteins in our benchmarking dataset to inform our ability to set thresholds between broad structural similarity and true structural mimicry. When this pipeline is applied to many parasite proteins, we’d expect to see many examples of structural similarity between parasite and human proteins that aren't “true” structural mimicry. Thus, it's critical that we include examples of this in our methods development to inform the thresholds we set for determining true mimicry.
What is “true” structural mimicry?
We define a “true” structural mimic as a parasite protein with a structure sufficiently similar to a human protein to have some of the same binding partners or specific substrates. We established this definition because it best meets our goal to use structural mimicry as a pointer to make mechanistic hypotheses about parasite modulation of human immune biology. The definition doesn’t work for other types of protein mimicry, like linear antigenic mimicry (e.g., 10) or purely functional mimicry (e.g., 11), but that’s not what we’re looking for with this particular search.
Goals and questions
Our overarching goal in building this pipeline was to use parasite structural mimicry to identify new ways to modulate the human immune system. We built this pipeline such that it could scale across all human-infecting viruses, as well as other human parasites.
Our key questions going into this project were:
- Pipeline development: What’s the best way to identify a viral mimic computationally and statistically?
- Interpreting results: How can we distinguish true viral mimicry of a specific human protein from the presence of broadly shared structural domains common to humans and viruses?
We’ve answered our first question, as our pipeline successfully identifies experimentally validated mimics. When we compare the strength of structural relationships between well-studied mimics to their human counterparts, however, we find that they have a wide range of structural similarity that overlaps with the range of structural similarity we see in broadly shared structural domains. Instead of implementing a hard threshold, we recommend that the user set their own thresholds based on what type of relationships they're trying to discover and their tolerance for false positives or false negatives. We’ve included an interactive plot for readers to play around with different thresholds to see how that impacts the types of results returned.
Our strategy
To build a structural mimicry detection pipeline, we needed to decide on which structural databases to use, select software and search parameters for detecting structural similarity, and implement a statistical method for selecting hits. We decided to use Viro3D 12 as our source of viral protein structures, and AlphaFoldDB 13 for our human structures. We ultimately decided on using Foldseek 3Di+AA 14 to do structural comparisons and Bayesian Gaussian mixture modeling (GMM) to cluster top candidates. A short breakdown of how these steps fit together in our pipeline can be found below in (Figure 2), and you can read on to the methods section for a detailed description of our full pipeline and decision-making process.
Briefly, the pipeline has the following steps:
- Download relevant predicted viral protein structures from Viro3D 12 and all predicted human structures from AlphaFoldDB 13. For the viral structures, also download the precomputed cluster information from Viro3D (based on sequence and structure).
- Compare each viral protein structure against all human protein structures using Foldseek 14.
- Perform Bayesian Gaussian mixture modeling (GMM) to cluster top candidate matches between human structures and groups of related viral protein structures.
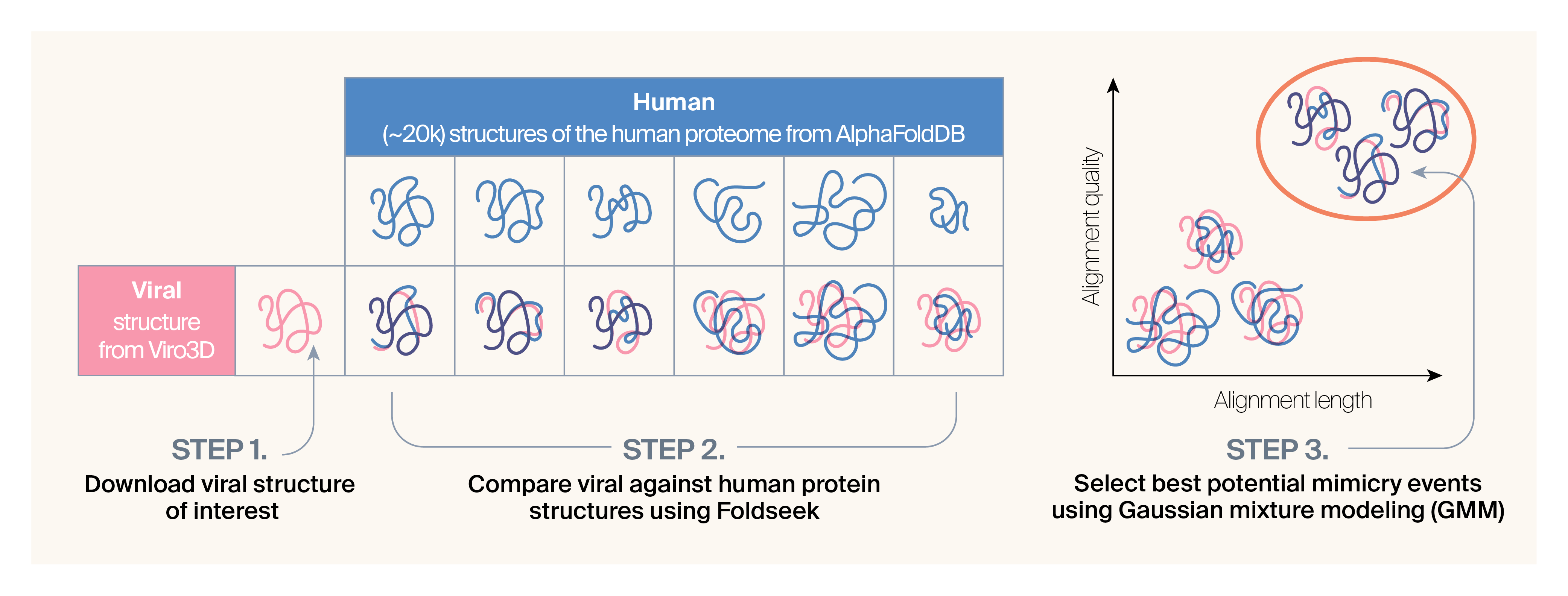
Figure 2. Overview of methods and data covered in this pub.
In this figure, we show our approach for a single viral protein. In addition to downloading the structure from Viro3D, we also retrieve clustering information from Viro3D. We run Gaussian mixture modeling (GMM) on Foldseek matches from a single viral cluster at a time.
The method
This is a detailed description of the pipeline that we built, as well as our considerations in making the decisions we did. We’ve also called out questions that came up as we were developing this approach in case other readers have answers. If you have thoughts on our method or answers to the questions we pose, please add them as comments so other readers and users can benefit!
Curating computationally predicted structures of viral benchmarking proteins and host proteins
For method development, we chose to focus on viruses that infect humans, as structural mimicry of human-infecting viruses has been studied for decades. To do our analysis, we used predicted human protein structures from AlphaFold 13 and predicted viral structures from Viro3D 12. Viro3D folded proteins using two methods (ColabFold 15 and ESMFold 16) and we used the structure with the higher quality score (pLDDT). In most cases, this was the ColabFold structure.
The viral structures we used in this analysis are available in our GitHub repository.
Below is the list of viral proteins we used to benchmark our approach. We began by curating well-studied examples from published reviews of parasite mimicry 2, 6, then expanded the list through a deeper literature review (Table 1). During this process, we identified a few viral proteins labeled in the literature as mimics based not on similarity to a single host protein but on shared structural features with many human proteins (Table 2). We included these incompletely characterized mimics in our benchmarking because we expect to encounter similarly ambiguous and even less well-characterized mimics in future, expanded analyses. However, a key question from the outset was whether these are legitimate mimics or simply represent domains that are broadly conserved across humans and viruses.
We also added two viral proteins (Table 3) not previously described as mimics in the literature, but which we suspected might fall into a "twilight zone" of similarity. We selected viral helicases and kinases based on the expectation that they’d have some baseline similarity to their ubiquitous counterparts in the human proteome.
| Viral protein (links to Viro3D) | Viral structure pLDDT | Viral species | Mimicked human protein (links to UniProt) | Human structure pLDDT | Reference* |
|---|---|---|---|---|---|
| BHRF1 | 84.4 | Epstein–Barr virus | Bcl-2 | 73.6 | 17 |
| BALF1 | 74.2 | Epstein–Barr virus | Bcl-2 | 73.6 | 18 |
| D19L (similar to C1L) | 72.7 | Vaccinia virus | Bcl-2 & PYDC1 | 73.6 & 88.8 | |
| CPXV036 (similar to C1L) | 69.9 | Vaccinia virus | Bcl-2 & PYDC1 | 73.6 & 88.8 | |
| VACWR027 (similar to C1L) | 51.6 | Cowpox virus | Bcl-2 & PYDC1 | 73.6 & 88.8 | 19 |
| CPXV034 | 93.1 | Cowpox virus | C4BP | 81.8 | |
| VACWR025 | 92.7 | Vaccinia virus | C4BP | 81.8 | 20 |
| D12L | 92.5 | Variola virus | C4BP | 81.8 | |
| US28 | 83.5 | Human cytomegalovirus | CCR1 | 82.8 | 21 |
| ORF74 | 79.9 | Kaposi’s sarcoma-associated herpesvirus | CXCR2 | 78.6 | |
| VACWR162 | 82.5 | Vaccinia virus | CD47 | 86.4 | 22–23 |
| 128L | 74.9 | Yaba monkey tumor virus | CD47 | 86.4 | |
| Integral membrane protein (murmansk-155) | 83.8 | Murmansk poxvirus | CD47 | 86.4 | |
| VACWR034 | 91.1 | Vaccinia virus | eIF2α | 77.0 | 9 |
| 12L | 90.5 | Yaba monkey tumor virus | eIF2α | 77.0 | |
| B9R | 87.1 | Monkeypox virus | IFNγR1 | 66.0 | |
| VACWR190 | 86.6 | Vaccinia virus | IFNγR1 | 66.0 | 24 |
| Interferon-gamma receptor (AKMV-88-197) | 87.8 | Akhmeta virus | IFNγR1 | 66.0 | |
| UL111A | 76.6 | Human cytomegalovirus | IL-10 | 88.0 | |
| UL111A | 86.2 | Simian cytomegalovirus | IL-10 | 88.0 | |
| BCRF1 | 86.9 | Epstein–Barr virus | IL-10 | 88.0 | 25–26 |
| MC054L | 75.7 | Molluscum contagiosum virus | IL-18BP | 79.0 | 27 |
| 14L | 88.4 | Yaba monkey tumor virus | IL-18BP | 79.0 | |
| D5L | 87.3 | Variola virus | IL-18BP | 79.0 | |
| NMDA receptor-like protein (CMLV006; similar to cowpox S1R) | 90.6 | Camelpox virus | TMBIM4 | 92.6 | 28 |
| US21 | 93.2 | Human cytomegalovirus | TMBIM4 | 92.6 |
Table 1. Well-characterized viral mimics and their human protein matches.
*At least one viral protein per mimicked human protein is well characterized and experimentally validated, and thus has a reference.
| Viral protein (links to Viro3D) | Viral structure pLDDT | Viral species | Protein type | Reference |
|---|---|---|---|---|
| MC148R | 72.5 | Molluscum contagiosum virus | Chemokine | 29 |
| NSP16* | 90.4 | Human coronavirus HKU1 | RNA methylase | 30 |
| NSP16* | 92.1 | Severe acute respiratory syndrome coronavirus 2 | RNA methylase | 30 |
| NSP5 | 92.4 | Human coronavirus HKU1 | Protease | 30 |
| NSP5 | 93.2 | Severe acute respiratory syndrome coronavirus 2 | Protease | 30 |
Table 2. Incompletely characterized viral mimics.
*NSP16 is labeled as NSP13 in the Viro3D database. This protein encodes an RNA methylase (PFAM domain PF06460) as a product of replicase polyprotein 1ab (orf1ab) cleavage and is most commonly referred to as NSP16.
| Viral protein (links to Viro3D) | Viral structure pLDDT | Viral species | Protein type | Reference |
|---|---|---|---|---|
| N-terminal helicase domain of the DEAD-box helicase superfamily | 89.1 | Human pegivirus genotype 2 | Helicase | |
| BGLF4 | 87.2 | Epstein–Barr virus | Kinase | 31 |
Table 3. Viral proteins with common domains.
Selecting tool and parameter combinations for structural comparisons
Using the well-characterized viral mimics as ground truth, we evaluated structural comparison approaches to see which tools and parameters maximized our ability to recover correct hits while minimizing off-target hits. We evaluated 3Di+AA and TM-align modes in Foldseek (v9.427df8a) 14. Foldseek 3Di+AA uses a hybrid alignment approach that encodes 3D geometry and amino acid identity, while Foldseek TM-align mode uses a structural superposition approach based on backbone geometry 14, 32. We focused on Foldseek in particular because it enables rapid, large-scale comparisons, which should allow us to scale our approach to larger datasets. While Foldseek 3Di+AA is faster than Foldseek TM-align mode, it uses a local alignment approach, while TM-align is global 14. We weren’t sure which method would better detect shared structure between viral and host proteins, so we tested both.
For both methods, we chose the parameter combination we thought most likely to return the most accurate results for each of these tools: for TM-align mode, we set --tmalign-fast 0 to turn “fast mode” off. This disables Foldseek's fast approximation and runs full TM-align iterations, optimizing the TM-score through detailed alignment refinement and structural superposition for more accurate results. For TM-align mode and 3Di+AA mode, we set --exact-tmscore 1 to turn on exact TM-score calculation. This enables a full structural superposition and exact TM-score calculation using the final alignment, providing a more accurate measure of structural similarity than the default approximate method. Foldseek also provides a --tmscore-threshold parameter that enables the user to set a minimum TM-score that alignments must meet to be reported in the output. We set the threshold to 0.5, a standard cutoff for structural homology 33. Using these parameter combinations, we compared each selected viral protein structure against all human protein structures that had a file available for download on AlphaFold (n = 20,174).
Removing poor-quality alignments
When we examined our data, we found that 3Di+AA mode returned many short alignments compared to TM-align mode (Figure 3, A), and that many of these short alignments had very low query TM-scores (Figure 3, B). We removed these extremely low-quality 3Di+AA hits, keeping hits with an alignment length greater than 20 and a query TM-score greater than 0.15 (Figure 3, B).
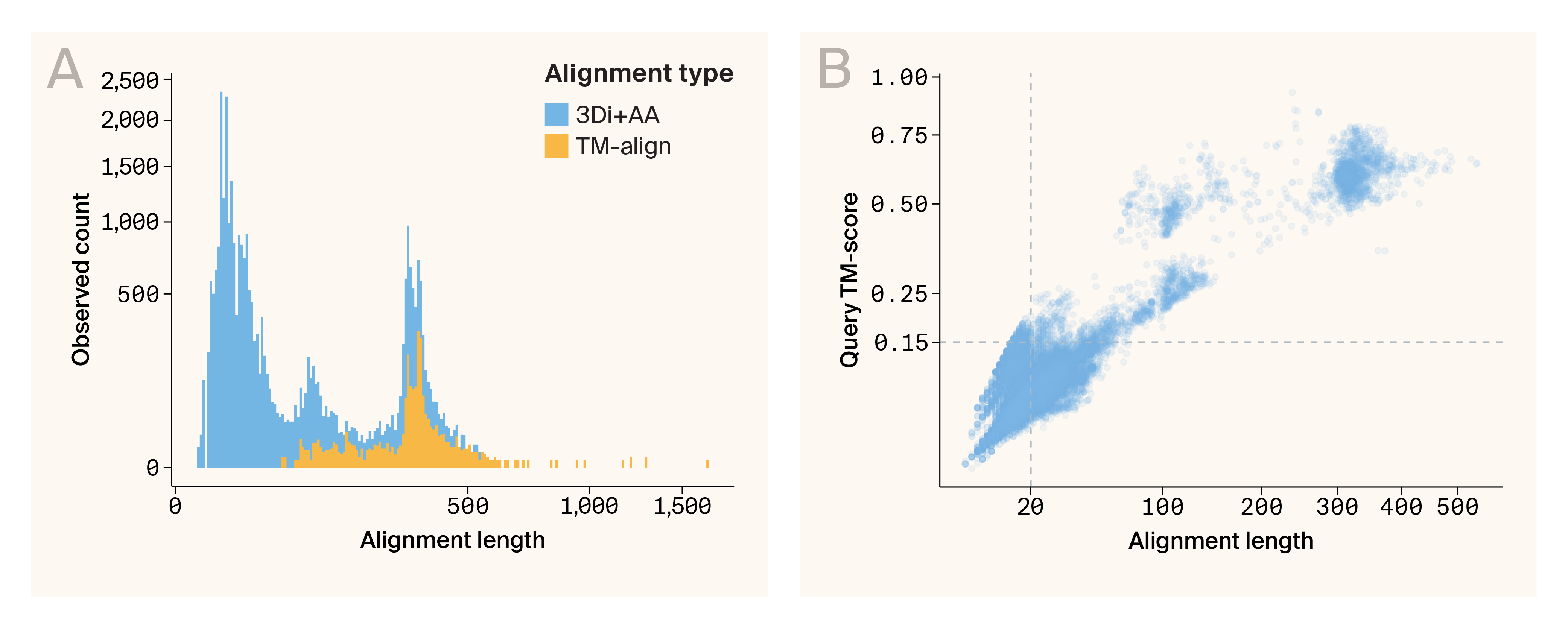
Figure 3. Foldseek 3Di+AA method returns many poor-quality alignments.
(A) Histogram comparing the number of alignments returned by Foldseek in 3Di+AA mode vs. TM-align mode. While the number of alignments returned above 100 amino acid residues long is comparable between the two methods, Foldseek 3Di+AA returns many short alignments.
(B) Scatter plot of alignment length by query TM-score of matches from Foldseek 3Di+AA. The dashed lines represent the filtering criteria we chose — a minimum alignment length of 20 and minimum query TM-score of 0.15. Matches must meet both requirements to be included.
Note on Foldseek thresholds
You might be wondering why the Foldseek 3Di+AA results include hits with query TM-scores far below the 0.5 prefiltering threshold that we implemented. This is because in the version of Foldseek we used (v9.427df8a), prefiltering thresholds use the alignment TM-score, not the query TM-score, to prefilter. Alignment TM-scores are normalized by the length of the aligned region, not the length of the full query protein. This means that proteins with alignments over extremely short regions are not filtered out. The latest version of Foldseek (v10.941cd33) allows users to prefilter on alignment, query, or target TM-score, but we haven’t tested it out yet.
Identification of mimicry events
For each benchmarking protein, we looked at alignment length (amino acid length of the structural match), query TM-score (structural similarity normalized by the length of the query viral protein), and the E-value (significance of hit, negative log-transformed in our figures). Foldseek TM-align and 3Di+AA modes both report alignment length and query TM-score, but only E-value calculations from 3Di+AA are meaningful. E-values reported from TM-align mode are actually TM-scores instead of E-value calculations (at least in Foldseek v9.427df8a; see this GitHub issue), so we've omitted them from Figure 4 14.
Open question
We wonder if it’s possible to derive an E-value for Foldseek results generated in TM-align mode. If so, what method or equation would be most appropriate?
When we look at the distributions of scores for each viral mimic, we find that the true match receives high query TM-scores and comparatively low E-values (which appear as high scores when negative log-transformed) (Figure 4). However, we also noticed cases where the true match scored well, but wasn’t the top hit for every metric (e.g., the Bcl-2 1 true match has the strongest E-value, but not the highest query TM-score). Also, the scores of the true matches were often nearly indistinguishable from the scores of off-target hits (see IL-10 2 and TMBIM4). In some cases, the true match wasn’t recovered at all (IL-10 1). Last, viral proteins are known to mimic multiple human proteins 19, necessitating a method that can return more than one human protein as a potential match.
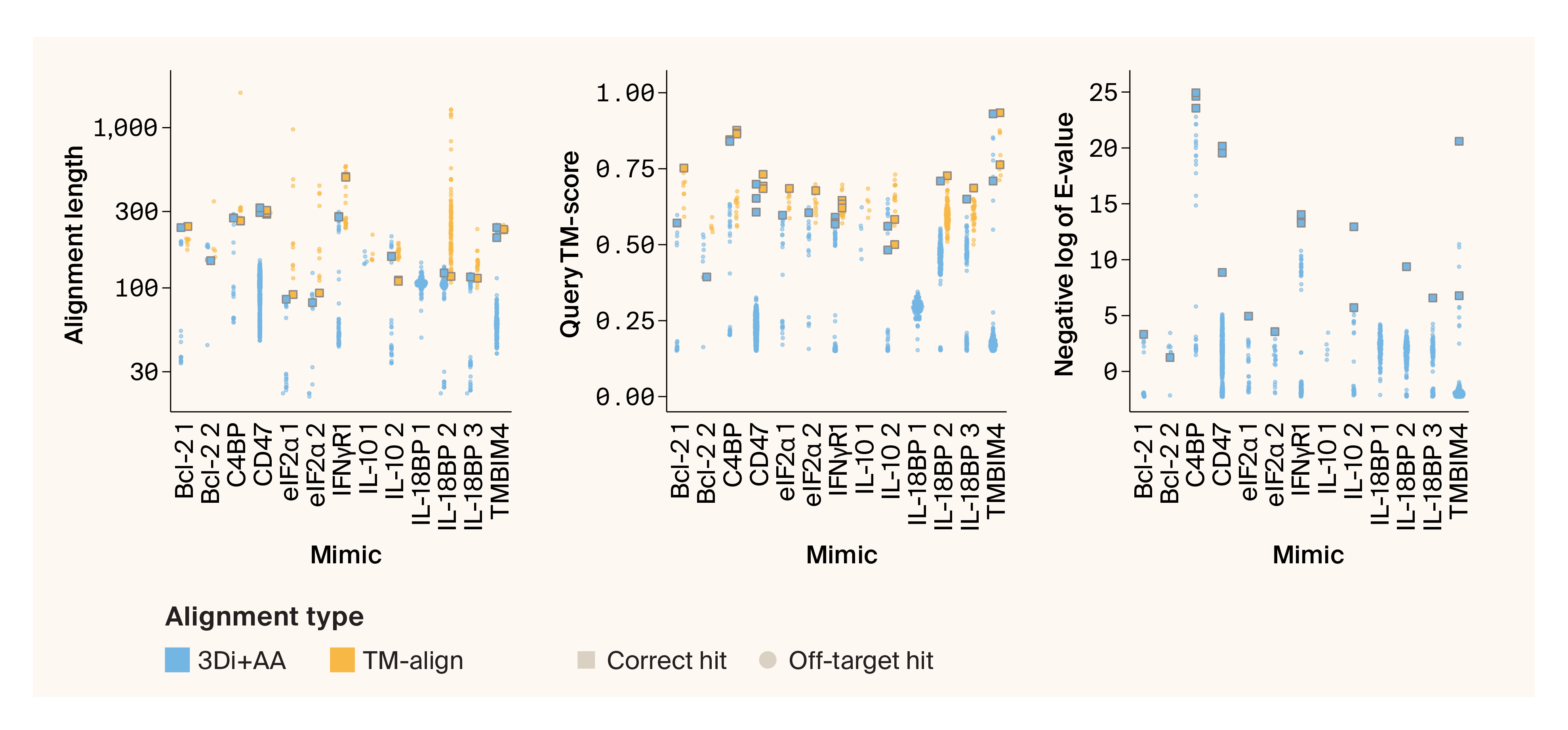
Figure 4. Distributions of TM-align and 3Di+AA scores for well-characterized mimics.
Quasi-random beeswarm plots illustrating the distribution of Foldseek hits from 3Di+AA and TM-align modes. Only 3Di+AA returns a meaningful E-value, so we’ve omitted TM-align E-values from the third panel.
Correct hits are depicted in squares, while off-target hits are shown as dots. The x-axis is labeled with the name of the human protein our viral query proteins mimic, as well as a numerical differentiator for Viro3D clusters when there are multiple.
Overall, this potential for complexity left us concerned that simply reporting the top hit for each viral protein would be misleading. So instead of choosing one metric (E-value or query TM-score) and assigning each viral protein its top hit as a potential host counterpart, we decided to implement a method to identify statistically distinguishable clusters of best hits, which we could then follow up by more carefully analyzing the individual scores for a given hit and examining the viral-host protein structural alignment.
Building a clustering framework with GMMs
To find clusters of top hits for each protein, we ultimately settled on Bayesian Gaussian mixture modeling (GMM). GMM is a probabilistic modeling approach that can use multiple types of data to identify underlying clusters of similar points within a complex dataset 34. We also chose to apply our modeling approach to clusters of viral proteins that had similar structures, instead of treating each viral protein individually. We’re assuming that structurally similar viral proteins likely mimic the same host protein, so doing our analysis on the level of viral clusters instead of individual proteins can give us more detection power. Viro3D has precomputed clusters for all viral protein structures (hereafter referred to as “Viro3D clusters”) 12, and we used these precomputed clusters for our downstream analysis.
Having calculated structural comparisons using Foldseek’s TM-align and Foldseek 3Di+AA modes, we wanted to test which dataset would result in better clustering and mimic identification. We decided to directly compare the performance of these different datasets in the GMM framework to identify mimicry. To do this, we built GMMs using E-value, query TM-score, and alignment length for well-characterized mimics. We compared three different models built from different underlying datasets:
- 3Di+AA: Foldseek 3Di+AA E-value, query TM-score, and alignment length.
- Hybrid: Foldseek 3Di+AA E-value and Foldseek TM-align query TM-score and alignment length.
- TM-align: Foldseek TM-align query TM-score and alignment length.
For the models that incorporate E-values (3Di+AA and hybrid), we selected the clusters that had the lowest mean E-value as the top-scoring clusters. For the TM-align model, we used the highest mean query TM-score to define the top-scoring cluster. In both cases, if fewer than 10 hits were returned, we didn't perform clustering but instead considered all hits as members of the same “best” cluster.
The viral protein query structures we used in this work and code for processing the Foldseek search results, running Gaussian mixture models, and creating the figures for the pub are available in our GitHub repo (DOI: 10.5281/zenodo.15398297).
Selecting the best model for mimicry detection
We evaluated how each of our models (3Di+AA, hybrid, and TM-align) performed in identifying the correct targets of well-characterized mimics. Our two points of evaluation were 1) how well each approach did in identifying mimicked human proteins, and 2) how many off-target hits each method returned. We found that the 3Di+AA model was able to identify 11 out of 11 mimicked host proteins (see details in Figure 5, and a summary in Figure 6). This model had an intermediate off-target rate. The hybrid model found 10 of 11 mimicked host proteins, but failed to match the viral C1L-like proteins (D19L, CPXV036, and VACWR027) to either of the two human proteins they're known to mimic — Bcl-2 and PYDC1, though it did identify other instances of Bcl-2 mimicry. That said, the hybrid model had the lowest off-target rate. The TM-align method performed the worst, finding 9/11 mimicked host proteins; it failed to match viral C1L-like proteins to either of the two human proteins they're known to mimic and failed to correctly match IFNγR1 mimics. It also had the highest off-target rate.
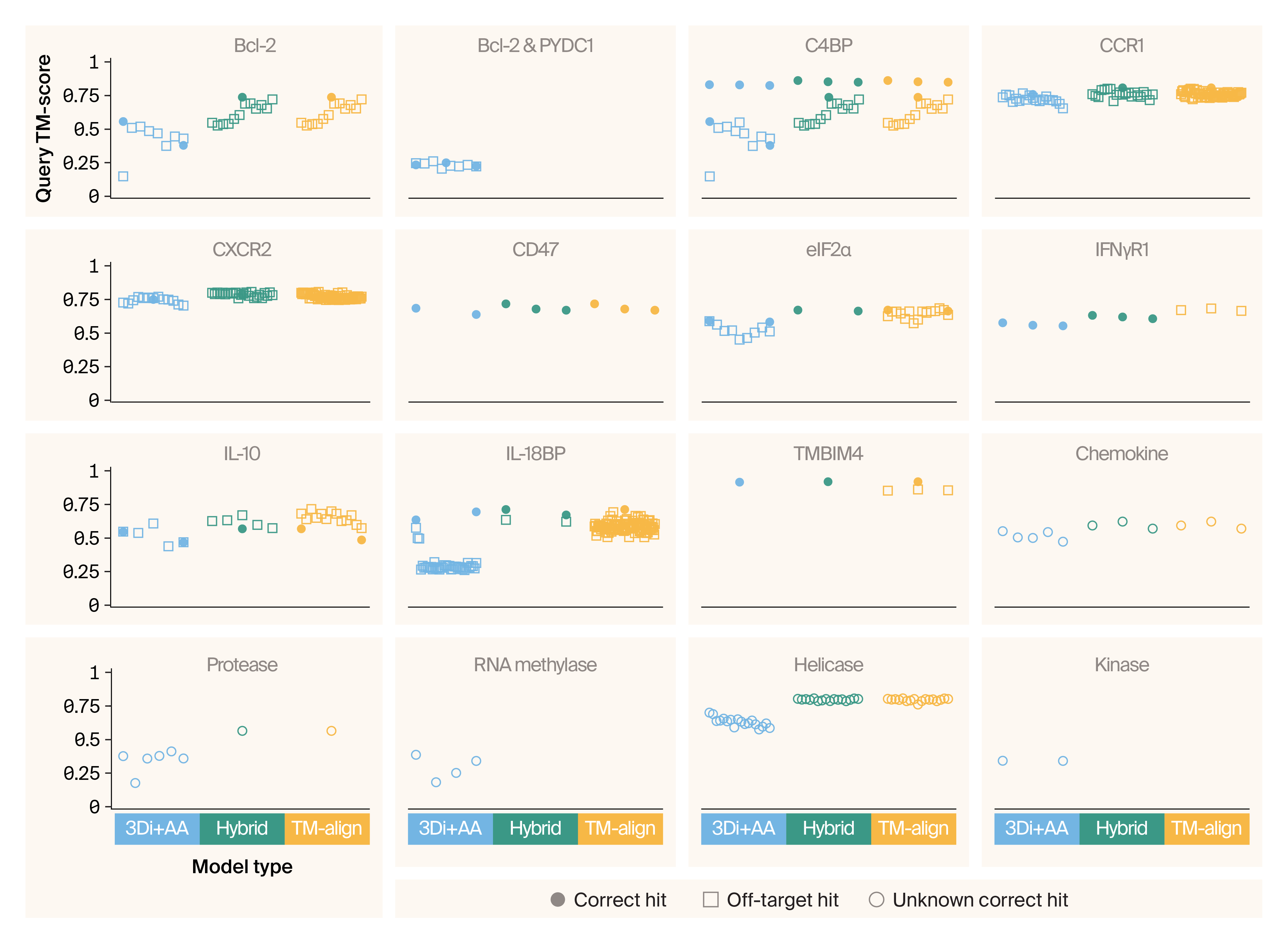
Figure 5. GMM applied to Foldseek 3Di+AA results accurately detects viral protein structural mimicry.
Jitter plot of correct, off-target, and unknown correct hits for controls mimics using measurements from Foldseek 3Di+AA alone, a hybrid of Foldseek 3Di+AA and TM-align mode, and TM-align mode alone. Click here to open an interactive version in a new tab. Hover over a point for details, including human & viral gene info.
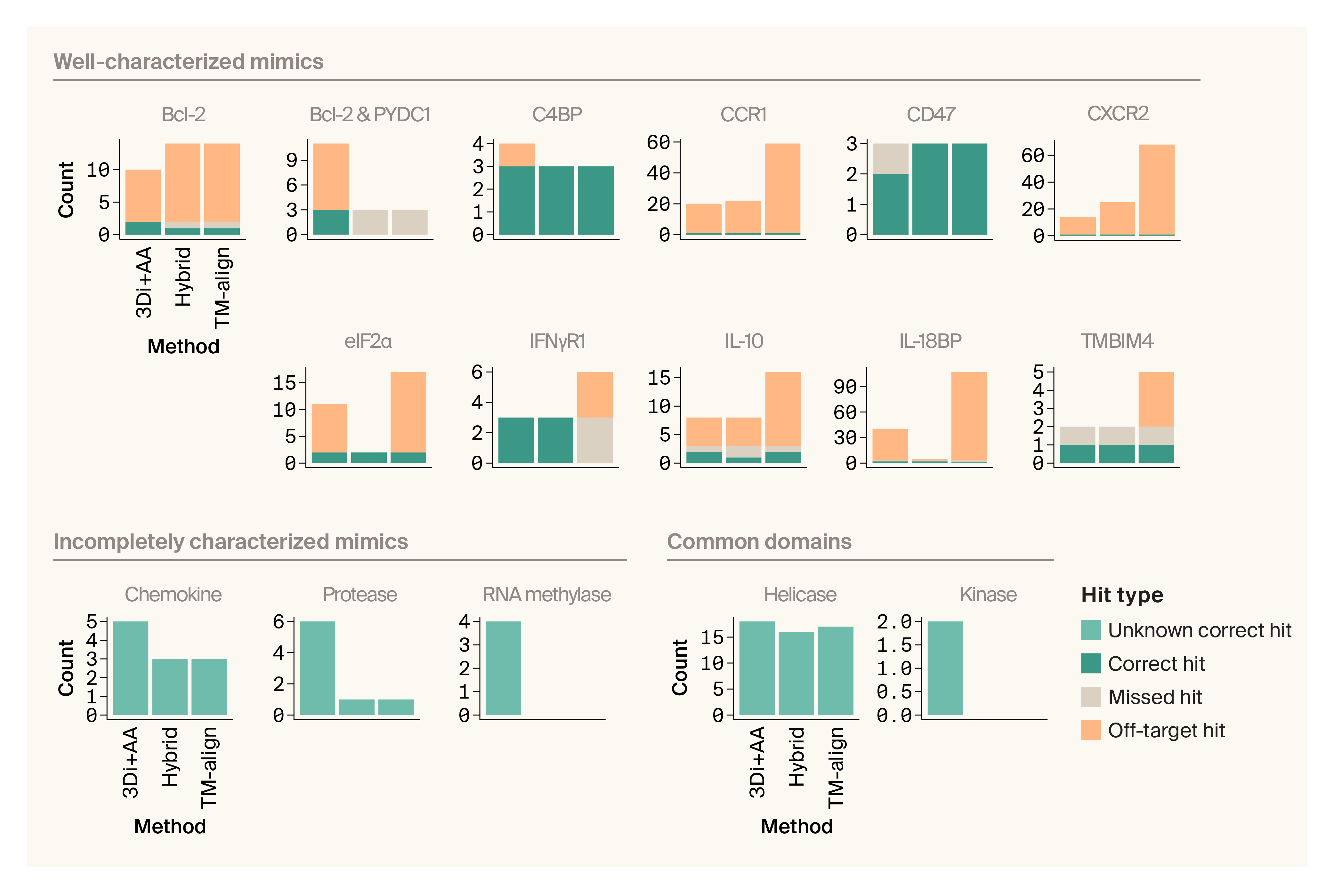
Figure 6. Foldseek 3Di+AA produces the most correct matches with few off-target hits for well-characterized viral mimics.
Bar plots counting the number of correct, missed, off-target, and unknown correct hits for different benchmark proteins.
We also looked at what happened with the incompletely characterized viral mimics (grouped by domain, and referred to here as chemokine, protease, and methylase). We didn’t have any strong priors on how the models needed to perform, as it’s an open question as to whether these are true mimics or are simply broadly conserved domains. We found that Foldseek 3Di+AA recovered the most hits for these proteins compared to the other two models, and the protease and methylase domain proteins had low query TM-scores (Figure 5). In contrast, all methods returned intermediate-scoring hits for the chemokine mimic (Figure 5).
Similarly, for the benchmarking proteins we included that have common domains and no suggested mimicry in the literature (referred to here as helicase and kinase), we saw mixed results. We found that Foldseek 3Di+AA returned the most hits for the viral kinase, but saw that the query TM-score was quite low for these hits (Figure 5). All methods returned intermediate-scored hits for the helicase.
We decided to move forward with the 3Di+AA approach because it had the highest true-positive rate and an intermediate false-positive rate. As an additional benefit, 3Di+AA is also the fastest method to run, enabling subsequent searches at scale.
Open question
Are there other statistical frameworks or further improvements that others could consider if they want to improve this pipeline?
Tuning thresholds for high-confidence mimicry detection
When we plot the strength of structural relationships (under the 3Di+AA model) between well-characterized mimics, incompletely characterized mimics, and common domains, we see substantial overlap between these categories. Instead of implementing hard cutoffs for defining true mimicry, we’d recommend that the user set their own thresholds based on their own research questions and their tolerance for false positives vs. false negatives. You can use the interactive plot below to select different E-value and query-TM scores as cutoffs and see how they affect the results. You can submit your selection and reasoning through the plot as well, and can check this Airtable link to see what other readers thought would be reasonable cutoffs.
If you have more questions about a specific protein, see the detailed results we provide for each one in the following subsections. We’ve called out some protein-specific questions that came up for each of these subsections in case any readers have answers.
Share the cutoffs you’d select to identify cases of viral structural mimicry.
Well-characterized viral mimics are labeled by the human protein they mimic, while incompletely characterized mimics and viral proteins with common domains are labeled by protein type.
Correct hits are highlighted with filled-in circles, off-target hits with empty circles, and hits for incompletely characterized mimics/proteins with common domains with filled-in squares.
Instructions: Select the E-value (negative log-transformed, x-axis) and query TM-score (y-axis) cutoffs that you would use to identify mimicry. With your submission, please leave a comment explaining why you chose those cutoffs.
Click here to view a static version of this plot.
Results: Check out other readers’ cutoffs and reasoning here.
Additional methods
We used Gemini to help write code, clean up code, and troubleshoot the interactive scatter plot figure. We used Claude and ChatGPT to help write code, clean up code, add comments to our code, and suggest wording before choosing which small phrases or sentence structure ideas to use.
Detailed results for benchmarking proteins
Data from this pub, including our Foldseek search results and the selected potential mimicry events, is available on Zenodo.
In the sections below, we walk through how our pipeline performed on well-characterized mimics, incompletely characterized mimics, and viral proteins with common domains. For well-characterized mimics, we discuss whether the pipeline correctly assigned them to their true host counterpart, and if not, why. For incompletely characterized mimics and viral proteins with common domains, we talk through how they performed in our analysis, and share our interpretation of those results.
In each subsection, we include structural alignments to give you a sense of the overall structural similarities between the viral proteins we analyzed and the human proteins to which we compared them. For well-characterized mimics and their human counterparts, we show a representative viral mimic structure aligned to the human protein it’s known to mimic. For incompletely characterized mimics and viral proteins with common domains, we show representative viral protein structures aligned to the human protein that our pipeline determined to be the closest match.
Results for well-characterized benchmarking proteins
Below are the results of benchmarking our pipeline against high-confidence, well-characterized viral mimics (also compiled with key info in Table 1). We’ve grouped them by the human protein that they mimic. We’re overall happy with how our pipeline performed here because it correctly matched at least one viral mimic to each of the 11 human proteins we know to be targets of mimicry. It’s exciting that this approach is able to rediscover many of these relationships in a single analysis. However, we still think we can learn from the instances where we missed a mimic, and have called out our specific questions about this in the following subsections. We also show the structural alignments and GMM results for each structural cluster of well-characterized mimics.
Mimicry of human Bcl-2 by viral proteins BALF1 and BHRF1
Human Bcl-2 aligned with viral protein BHRF1.
Predicted Bcl-2 is blue, predicted BHRF1 is pink. Aligned with the PyMol CE algorithm.
Human protein function: Apoptosis regulator Bcl-2 is a pro-survival protein that suppresses apoptosis 35.
Human protein superfamily: Bcl-2 is part of the Bcl-2 inhibitors of programmed cell death superfamily (SSF56854). There are at least 19 proteins in this superfamily encoded in the human genome 36.
Prediction of viral mimicry: The Epstein–Barr herpesvirus encodes multiple proteins that mimic Bcl-2. Both BHRF1 and BALF1 have structural and sequence similarity to human Bcl-2 17–18, 37–38.
Experimental evidence of mimicry: The BHRF1 protein inhibits apoptosis by binding to known human Bcl-2 interactors such as Bim and other pro-apoptotic proteins 8, 39. The role of BALF1 is less clear, with conflicting findings suggesting both pro- and anti-apoptotic functions 18, 38, 40.
Our results: The two query proteins were in two different Viro3D clusters. For BHRF1, the GMM we ran returned human Bcl-2 as its top hit. For BALF1, Foldseek only returned nine hits, so we didn’t run any modeling but instead kept all hits. These included Bcl-2 as well as seven other Bcl-2 homologs and a non-homolog protein, IZUMO2. Bcl-2 wasn't the top hit, however — MCL1 is the top hit by E-value. Overall, this matches experimental evidence of BHRF1 being a clear apoptosis inhibitor while BALF1 has recognizable homology to human proteins in the Bcl-2 superfamily but unclear function.
GMM output: We’ve shared interactive plots with GMM clustering of Foldseek structural comparison results for the viral BHRF1 protein here and the viral BALF1 protein here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Open question
Since Bcl-2 refers to a protein and a family of proteins, it’s unclear whether BALF1 hitting Bcl-2 homologs represents our inability to recover the true hit or whether BALF1 mimics one of these proteins. We'd be curious to hear which scenario is more likely from experts who study Bcl-2 mimicry.
Mimicry of human proteins Bcl-2 and PYDC1 by the viral fusion proteins D19L, CPXV036, and VACWR027
Human PYDC1 aligned with the N-terminal domain of viral protein VACWR027.
Predicted PYDC1 is blue, predicted VACWR027 N-terminus is pink. Aligned with the PyMol CE algorithm.
Human Bcl-2 aligned with the C-terminal domain of viral protein VACWR027.
Predicted Bcl-2 is blue, predicted VACWR027 C-terminus is pink. Aligned with the PyMol CE algorithm.
Human protein function: Apoptosis regulator Bcl-2 is a pro-survival protein that suppresses apoptosis by binding to different proteins 35. Pyrin-domain-containing protein 1 (PYDC1) is a regulatory protein that inhibits inflammation by interfering with inflammasome assembly and caspase-1 activation 41.
Human protein superfamily: Bcl-2 is part of the Bcl-2 inhibitors of programmed cell death superfamily (SSF56854). There are at least 19 proteins in this superfamily encoded in the human genome 36. PYDC1 is part of the DEATH domain superfamily (SSF47986). There are at least 105 proteins in this superfamily encoded in the human genome 36.
Prediction of viral mimicry: A computationally predicted structure of C1L has structural homology with both Bcl-2-like proteins as well as pyrin-domain-containing proteins. The two globular domains of C1L are joined by a flexible linker 19.
Experimental evidence of mimicry: Unlike other poxvirus Bcl-2 mimics and human Bcl-2, the C1L Bcl-2 domain is not anti-apoptotic 42. Instead, both domains of the C1L protein interact with the host ASC protein to promote ILβ-mediated inflammasome signaling 19. While this is a new functional role for a Bcl-2 mimic, this is similar to the role of some host pyrin-domain-containing proteins.
Our results (full-length): We queried with three poxvirus proteins with homology to C1L. All three were in the same Viro3D cluster. All three returned PYDC1 (query TM-score range 0.21–0.28) and other pyrin-domain-containing proteins, reflecting the presence of this domain in the fusion proteins. No protein matched against Bcl-2 or homologous proteins. We wondered if decomposing C1L into its two domains would improve our ability to detect the Bcl-2 domain, but that didn’t work (see below). The authors of the study 19 that identified the Bcl-2 domain used FATCAT 43 as their structural aligner instead of Foldseek, which may underlie these differences in detection.
GMM output (full-length): We've shared an interactive plot with GMM clustering of Foldseek structural comparison results for full-length viral C1L-like proteins here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Open question
Are there other high-throughput approaches to scan for fusion proteins that contain two or more domains that represent protein structural mimicry?
Our results (split proteins): In addition to querying with the entire protein structure, we split each protein into its constituent domains. We wanted to know whether our approach could detect each domain individually. When we queried with the pyrin-domain-containing domain, we didn't return PYDC1 as above, but did return hits to other pyrin-domain-containing proteins [PYDC2, NLRP3, NLRP4, NLRP6, NLRP11, and NLRP13 (nucleotide-binding oligomerization domain, leucine-rich repeat, and pyrin-domain-containing)]. When we queried with the Bcl-2-domain-containing domain, we only saw an off-target hit to striatin-4. This hit was the best match, but also had a very low query TM-score (0.18) and poor E-value (32), suggesting this is not a hit that represents true mimicry. We aren’t sure why we didn’t recover Bcl-2 hits, given C1L’s annotation as a Bcl-2-like protein.
GMM output (split proteins): We've shared interactive plots with GMM clustering of Foldseek structural comparison results for the viral PYDC1-like domains here and Bcl-2-like domains here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Open question
C1L is annotated as a Poxvirus_Bcl-2-like domain protein. Is it surprising that poxviral Bcl-2-like domain proteins are highly structurally divergent from human Bcl-2 proteins?
Mimicry of human TMBIM4 by viral proteins CMLV006 and US21
Human TMBIM4 aligned with viral protein CMLV006.
Predicted TMBIM4 is blue, predicted CMLV006 is pink. Aligned with the PyMol CE algorithm.
Human protein function: Protein lifeguard 4 (TMBIM4, historically Lfg4), also referred to as Golgi anti-apoptotic protein (GAAP) and transmembrane BAX inhibitor motif containing 4, is a protein that localizes to the Golgi apparatus and confers resistance to apoptotic stimuli inside and outside the cell 28, 44–45.
Human protein superfamily: TMBIM4 is part of the Bax inhibitor superfamily. There are at least eight proteins in this superfamily encoded in the human genome 36.
Prediction of viral mimicry: The viral TMBIM4-like protein encoded by camelpox virus protein 6L has approximately 73% sequence similarity to human TMBIM4 28. Both the vaccinia virus TMBIM4-like protein (called v-GAAP in this publication and others) and camelpox virus v-GAAP proteins have a conserved architecture, which is supported by epitope tagging and selective membrane permeabilization studies 46.
Experimental evidence of mimicry: The viral TMBIM4-like proteins (vaccinia virus strain Evans v-GAAP and camelpox virus strain CM-S v-GAAP) inhibit apoptosis in a similar way to human TMBIM4 28. The function of the two proteins overlaps enough that when human TMBIM4 is knocked out, viral TMBIM4-like proteins (vaccinia virus strain Evans v-GAAP and camelpox virus strain CM-S v-GAAP) can substitute for it and prevent cell death 28.
Our results: We used two viral proteins to test for mimicry of TMBIM4 — an experimentally validated camelpox protein 28 and a homologous cytomegalovirus protein US21. Both proteins were in the same Viro3D cluster, so we only ran GMM once. This only returned TMBIM4. However, while both proteins have Foldseek matches to TMBIM4, the camelpox protein match was so much stronger that the cluster we selected from the model only contained the camelpox protein. This is potentially both a pro and a con of our method — we recovered the strongest hit, but our strong hit essentially “outcompeted” another valid hit. In this case, actually looking at the clustering graph is very helpful for uncovering this behavior.
GMM output: We've shared an interactive plot with GMM clustering of Foldseek structural comparison results for viral TMBIM4-like proteins here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Mimicry of human CCR1 by viral protein US28
Human CCR1 aligned with viral protein US28.
Predicted CCR1 is blue, predicted US28 is pink. Aligned with the PyMol CE algorithm.
Human protein function: Human C-C chemokine receptor type 1 (CCR1) triggers a signaling cascade in immune cells that leads to migration toward the chemokine source when the receptor binds its ligands CCL3, CCL5-9, CCL13-16, and CCL23 47.
Human protein superfamily: CCR1 is part of the family A (rhodopsin family) G-protein-coupled receptor-like superfamily (SSF81321). The human genome encodes at least 948 proteins in this superfamily 36.
Prediction of viral mimicry: The human cytomegalovirus protein US28 encodes a chemokine receptor with homology to human CCR1, CCR5, and CX3CR1 48–50. While the cytomegalovirus likely obtained US28 via horizontal transfer of a GPCR from a host, crystal structures of protein US28 in complex with chemokine ligands show a different binding mechanism from human chemokine receptor–ligand binding 51.
Experimental evidence of mimicry: The US28 protein mimics CCR1 but displays substantially expanded functionality. US28 binds the human CCR1 ligands as well as those of CCR5 and CX3CR1 (CCL1, CCL2, CCL3, CCL4, CCL5, and CX3CL1) 49–50, 52–54. Ligand binding induces intracellular signaling, but the form this takes depends on the bound chemokine and the infected cell. For example, in smooth muscle cells, CC chemokines promote migration, while CX3CL1 blocks migration 55–56. In contrast, in macrophages, CX3CL1 induces migration, while CCL5 inhibits it 55, 57–58.
Our results: Our US28 query against the human proteome returned many chemokine receptors (CCR1–CCR5, CCR7–CCR10, CXCR1, CXCR3–5, XCR1, CX3CR1), including two atypical chemokine receptors (ACKR2, ACKR1). It also returned receptors from other classes, including two bradykinin receptors (BDKRB1, BDKRB2) and one angiotensin receptor (AGTR2). These results encompass the three human receptors to which US28 has documented homology (CCR1, CCR5, and CX3CR1 48–50) as well as additional proteins. A scatter plot of Foldseek query TM-score, alignment length, and E-value for US28 results shows that while the model selected many hits, not all are equally strong — CX3CR1 stands out, consistent with its known relationship to US28.
GMM output: We’ve shared an interactive plot with GMM clustering of Foldseek structural comparison results for the viral US28 protein here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Mimicry of human CXCR2 by viral protein ORF74
Human CXCR2 aligned with viral protein ORF74.
Predicted CXCR2 is blue, predicted ORF74 is pink. Aligned with the PyMol CE algorithm.
Human protein function: Human C-X-C chemokine receptor type 2 (CXCR2) activates intracellular signaling pathways that promote chemotaxis, inflammation, and recruitment of neutrophils to sites of infection or injury when the receptor is bound by its agonists CXCL1–3 and CXCL5–8 59.
Human protein superfamily: CXCR2 is part of the family A (rhodopsin family) G-protein-coupled-receptor-like superfamily (SSF81321). The human genome encodes at least 948 proteins in this superfamily 36.
Prediction of viral mimicry: Kaposi’s sarcoma-associated herpesvirus ORF74 encodes a G-protein-coupled receptor with some sequence homology to human IL-8 chemokine receptors CXCR1 and CXCR2 60, and structurally resembles CXCR2 61.
Experimental evidence of mimicry: ORF74 binds chemokines from both the CC and CXC families, while human CXCR2 only binds CXC chemokines 59. Also different from human chemokine receptors, ORF74 is constitutively active, activating proliferative and anti-apoptotic signaling pathways 62.
Our results: The ORF74 viral query returned 14 matches to chemokine receptors (CXCR1–CXCR4, CX3CR1, CCR3, CCR4, CCR7, CCR8, CCR10), atypical chemokine receptors (ACKR2–ACKR4), and an angiotensin receptor (AGTR1). This in part matches experimental evidence, as ORF74 has structural similarity to CXCR2 and sequence homology to CXCR1 and CXCR2 60–61. Matches to both CXC and CC chemokine receptors may also support ORF74’s ability to bind both CC and CXC chemokines 59. However, our approach returns additional chemokine receptors as well, which are of uncertain significance.
GMM output: We’ve shared an interactive plot with GMM clustering of Foldseek structural comparison results for the viral ORF74 protein here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Open question
Both viral chemokine receptors we used as queries return many hits, including to non-chemokine receptors. Is this expected behavior, or is our approach failing to capture a more precise set of mimicry candidates? Do the hits our method returns reflect what's known about each chemokine receptor mimic?
Mimicry of human CD47 by viral 128L, VACWR162, and murmansk integral membrane protein
Human CD47 aligned with viral protein VACWR162.
Predicted CD47 is blue, predicted VACWR162 is pink. Aligned with the PyMol CE algorithm.
Human protein function: Human cluster of differentiation 47 (CD47) is a transmembrane protein on the surface of many different cells in the body that functions as a “don’t eat me” signal so that macrophages or other immune cells don’t phagocytose “self” cells 63.
Human protein superfamily: CD47 is part of the immunoglobulin superfamily (SSF48726). The human genome encodes at least 1,188 proteins in this superfamily 36.
Prediction of viral mimicry: Poxvirus CD47-like proteins share 23–28% amino acid identity with mammalian CD47 proteins 23, 64.
Experimental evidence of mimicry: Both poxvirus CD47-like proteins and human CD47 localize to the cell membrane 65. When overexpressed, they both promote calcium influx and contribute to necrotic cell death via increased membrane permeability 22. Like human CD47, some poxvirus CD47-like proteins induce inhibitory signals in macrophages 65.
Our results: We queried the human proteome with three poxvirus proteins — yaba monkey tumor virus 128L, vaccinia virus VACWR162, and murmansk poxvirus integral membrane protein (Table 1). All three viruses were in the same Viro3D cluster, so we ran GMM once. While all three structures had real matches to CD47, our modeling approach returned only two hits, meaning that one viral CD47-like protein (yaba monkey tumor virus 128L) was overlooked because it has weaker similarity to CD47 than the others. Similar to our findings with TMBIM4 mimics, we found that the GMM selects the strongest hits, which can potentially exclude weaker, but legitimate, relationships. Looking at the scatter plot of E-value, query TM-score, and alignment length here is useful for finding overshadowed examples of real mimicry.
GMM output: We’ve shared an interactive plot with GMM clustering of Foldseek structural comparison results for viral CD47-like proteins here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Mimicry of human C4BP by viral proteins CPXV034, VACWR025, and D12L
Human C4BP aligned with viral protein VACWR025.
Predicted C4BP is blue, predicted VACWR025 is pink. Aligned with the PyMol CE algorithm.
Human protein function: C4-binding protein (C4BP) is a regulatory protein in the complement system that inhibits complement activation by binding to and inactivating C4b, thereby preventing the formation and stability of the C3 convertase enzyme complex 66–68.
Human protein superfamily: C4BP is part of the complement control module superfamily (SSF57535). The human genome encodes at least 49 proteins in this superfamily 36.
Prediction of viral mimicry: The vaccinia virus complement control protein C3L (VACWR025) contains four repeating motifs that are 60 amino acids long (common to proteins in the complement control module superfamily), and has an average of 33% amino acid identity to human C4BP 69. The human protein has eight complement control motifs, however, making the viral mimic markedly smaller.
Experimental evidence of mimicry: Like human C4BP, vaccinia virus complement-binding protein binds human C3b and C4b, blocking the complement cascade that would otherwise lead to virus neutralization 70–72.
Our results: We queried the human proteome with three poxvirus C4BP mimics: cowpox virus CPXV034, vaccinia virus VACWR025, and variola virus D12L. All three proteins were in the same Viro3D cluster, so we performed one modeling round. The top-scoring cluster included all three matches to C4BP; however, it also included one match to CD55 (another member of the complement control module superfamily). When we look at the scatter plot, we see that C4BP hits appear as a tight cluster separated from the CD55 match. When we look at the GMM probability of each protein belonging to the top-scoring cluster, we see that the C4BP hits have a higher probability of belonging to this cluster (all > 0.99) than the CD55 match (0.88). Overall, we find that our method returns expected relationships between proteins and that looking at the underlying data is helpful for refining hypotheses about mimicry.
GMM output: We've shared an interactive plot with GMM clustering of Foldseek structural comparison results for viral C4BP-like proteins here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Mimicry of human eIF2α by viral proteins VACWR034 and 12L
Human eIF2α aligned with viral protein VACWR034.
Predicted eIF2α is blue, predicted VACWR034 is pink. Aligned with the PyMol CE algorithm.
Human protein function: Human eIF2α is a critical regulator of protein synthesis that, when phosphorylated by PKR during viral infection, becomes inactivated, thereby halting translation initiation to suppress viral replication 73–75.
Human protein superfamily: The human eIF2α protein is part of multiple superfamilies, but the portion that is mimicked by viruses is part of the nucleic-acid-binding proteins superfamily (SSF50249). The human genome encodes at least 90 proteins in this superfamily 36.
Prediction of viral mimicry: Viral eIF2α mimics are small proteins that have sequence homology to a sub-region of eukaryotic eIF2α 76. Crystal structures of these viral proteins show that these proteins mimic the region of eIF2α that interacts with PKR (see next paragraph) 77.
Experimental evidence of mimicry: Viral eIF2α mimics are antagonistic proteins that create a decoy that PKR acts on 78. This allows the host eIF2α to remain unphosphorylated and for protein translation and viral replication to continue 9.
Our results: We queried with two eIF2α mimics from two poxviruses, each protein in a separate Viro3D cluster. The vaccinia virus protein encoded by VACWR034 matched to eIF2α alone. However, the yaba monkey tumor virus protein 12L matched against eIF2α as well as nine off-target matches. Most of these off-target matches are to other members of the nucleic-acid-binding proteins superfamily (SRBD1, PDCD11, EXOSC3, PNPT1, DIS3, ZCCHC17, EXOSC1). However, two off-target matches are outside of that family: DNA-directed RNA polymerase I subunit RPA43 (POLR1F) and threonylcarbamoyladenosine tRNA methylthiotransferase (CDKAL1). While eIF2α is technically the hit with the lowest E-value, we’d be unlikely to predict the function of the protein based on our mimicry analysis alone. We think this was a particularly challenging case for our approach — the viral eIF2α is a small, truncated mimic; it's 88 amino acids long and mimics less than half of the human protein.
GMM output: We've shared interactive plots with GMM clustering of Foldseek structural comparison results for the viral VACWR034 protein here and the viral 12L protein here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Open question
Are there other approaches we should think about that would be more appropriate for small, truncated mimics?
Mimicry of human IL-10 by viral proteins BCRF1 and UL111A (human and simian CMV)
Human IL-10 aligned with viral protein BCRF1.
Predicted IL-10 is blue, predicted BCRF1 is pink. Aligned with the PyMol CE algorithm.
Human protein function: Human interleukin 10 (IL-10) is a context-dependent cytokine that primarily suppresses immune responses by inhibiting monocytes, macrophages, and dendritic cells, but can also promote inflammation by activating B cells, stimulating mast cells, and supporting regulatory T cell differentiation 79–85.
Human protein superfamily: Human IL-10 is part of the four-helical cytokine superfamily (SSF47266). The human genome encodes over 86 proteins in this superfamily 36.
Prediction of viral mimicry: Epstein–Barr virus (gamma herpesvirus 4) mimics human IL-10 with its protein BCRF1 (vIL-10). BCRF1 shares high sequence identity with human IL-10 (84% in mature protein-coding sequence) 86–87. The BCRF1 crystal structure is similar to human IL-10 but has some novel conformations 25. In contrast, human cytomegalovirus UL111A shares 27% sequence identity with human IL-10 88 and has a similar structure 89.
Experimental evidence of mimicry: Like human IL-10, vIL-10 suppresses many host pro-inflammatory immune responses 90. However, conformational changes to the structure give BCRF1 reduced binding affinity to the human IL-10 receptor 1 26. This allows BCRF1 to avoid pro-inflammatory phenotypes of human IL-10, such as mast cell and thymocyte proliferation 91, because pro-inflammatory cell surfaces have reduced receptor expression on pro-inflammatory cell surfaces 92. In contrast, human cytomegalovirus UL111A shares similar binding affinity to human IL-10 receptor 1 as human IL-10 89.
Our results: We queried with three viral IL-10 mimics from the herpesvirus family (Table 1). These structures grouped into two Viro3D clusters, so we ran two rounds of GMM. Two IL-10 mimics, one encoded by the Epstein–Barr virus (BCRF1) and one by simian cytomegalovirus (UL111A), grouped in the same cluster. Our modeling approach returned only IL-10 for both viral proteins. In the second cluster, the Foldseek search with the human cytomegalovirus UL111A returned fewer than 10 proteins, so we didn’t run any modeling and instead kept all hits. However, none of these hits were to IL-10. The search instead returned IL-19, IL-20, IL-22, IL-24, and IL-26, which are all members of the same protein superfamily as IL-10. While these matches are similar to IL-10, we were surprised that we didn’t see IL-10 as a hit. Our best explanation right now is that the human cytomegalovirus IL-10 mimic UL111A has a lower-quality predicted structure than the two IL-10 mimics that successfully returned IL-10 (pLDDT of 76.6 vs. 86.2 and 86.9, respectively). It’s possible that the lower-quality structure reduced our ability to detect the true structural match for this protein. This highlights the importance of checking structure quality when interpreting results, and points out a limitation inherent to using predicted structures instead of experimentally determined structures.
GMM output: We've shared interactive plots with GMM clustering of Foldseek structural comparison results for viral BCRF1 and simian CMV UL111A proteins here and the human CMV UL111A protein here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Mimicry of human IL-18-binding protein by viral proteins MC054L, 14L and D5L
Human IL-18BP aligned with viral protein MC054L.
Predicted IL-18BP is blue, predicted MC054L is pink. Aligned with the PyMol CE algorithm.
Human protein function: Human interleukin-18-binding protein (IL-18BP) is a secreted decoy receptor that sequesters IL-18, an inflammatory cytokine 93.
Human protein superfamily: IL-18BP is part of the immunoglobulin superfamily (SSF48726). The human genome encodes at least 1,188 proteins in this superfamily 36.
Prediction of viral mimicry: The poxvirus molluscum contagiosum IL-18BP-like protein MC054L has 35% amino acid identity to human IL-18BP 94. Structural predictions of human and MC054L show that the protein has a conserved binding site for IL-18 94.
Experimental evidence of mimicry: Like human IL-18BP, the molluscum contagiosum IL-18BP mimic MC054L prevents IFNγ production in a dose-dependent manner 27. The vaccinia virus IL-18BP mimic C12L inhibits innate and adaptive immune responses typically coordinated by IL-18 during poxvirus infection, thereby achieving prolonged infection 95. The C12L protein also reduces natural killer cell cytotoxicity and cytotoxic T cell activity, increasing the length of infection 95.
Our results: We queried the human proteome with three poxvirus IL-18BP mimics — molluscum contagiosum MC054L, yaba monkey tumor virus 14L, and variola virus D5L. These proteins had the lowest similarity to each other of any of the mimics we tested and grouped into three separate Viro3D clusters. The yaba monkey tumor virus 14L protein returned IL-18BP alone. The variola virus D5L protein returned IL-18BP as well as three off-target hits (IL-1R2, CD200, NCR3LG1), all members of the same superfamily as IL-18BP. However, IL-18BP was an outlier among these hits, with the lowest E-value. The molluscum contagiosum MC054L returned 34 off-target hits, the majority of which were to proteins in the immunoglobulin superfamily. While experimental evidence supports that MC054L is indeed an IL-18BP mimic, unlike the human version, it also has an extended C-terminal tail that allows it to bind glycosaminoglycans 96. This may lead to the observed off-target hits.
GMM output: We've shared interactive plots with GMM clustering of Foldseek structural comparison results for the viral 14L protein here, the viral D5L protein here, and the viral MC054L protein here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Mimicry of human IFNγR1 by viral proteins B9R, VACWR190, and AKMV-88-197
Human IFNγR1 aligned with viral protein VACWR190.
Predicted IFNγR1 is blue, predicted VACWR190 is pink. Aligned with the PyMol CE algorithm.
Human protein function: Interferon γ receptor 1 (IFNγR1) binds interferon γ and triggers activation of the STAT1 transcription factor to initiate immune responses that enhance antiviral defense 97–98.
Human protein superfamily: IFNγR1 is part of the fibronectin type III superfamily (SSF49265). The human genome encodes at least 244 proteins in this superfamily 36.
Prediction of viral mimicry: The poxvirus Ectromelia virus IFNγR1-like protein C4R shares ~20% amino acid identity with the extracellular portion of human IFNγR1 99. The protein is also structurally similar to this portion of the human protein, as demonstrated by crystal structure comparisons 99.
Experimental evidence of mimicry: Poxvirus IFNγR1 mimics such as Ectromelia virus protein C4R and myxoma virus M-T7 bind human IFNγ 99–101. However, the viral version is a soluble decoy receptor instead of a membrane-anchored receptor protein 99–101. Poxviruses use the mimic to increase pathogenicity by dampening host IFNγ-mediated immune responses 101.
Our results: We queried with three poxvirus IFNγR1 mimics, monkeypox virus B9R, vaccinia virus VACWR190, and Akhmeta virus interferon-gamma receptor (AKMV-88-197), all of which belonged to the same Viro3D cluster. Our analysis only returned IFNγR1, which matches the existing experimental evidence for mimicry. Additionally, we hit all three viral proteins, indicating an equally strong match between all three query structures.
GMM output: We've shared an interactive plot with GMM clustering of Foldseek structural comparison results for viral IFNγR1 proteins here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Results for incompletely characterized mimics
In addition to the above examples of structural mimicry, we included viral proteins that have been described as mimics due to structural similarity to a human protein or class of protein, but for which a specific, well-validated human match isn’t known (key info listed in Table 2). Namely, we included a viral chemokine, protease, and methylase. We see that the viral chemokine has intermediate-scoring hits to human chemokines, and that the viral protease and methylase have sparse, low-scoring matches to human proteases and methylases, respectively. We interpret these results to mean that the chemokine is a true mimic, and the protease and methylase are both common domains. Below, we show the GMM clustering of matches as well as structural alignments of the viral proteins to the human protein to which they have the most structural similarity.
Mimicry of human chemokines by viral protein MC148R
Human CCL28 aligned with viral protein MC148R.
Predicted CCL28 is blue, predicted MC148R is pink. Aligned with the PyMol CE algorithm.
CCL28 was the hit with the lowest E-value in the best cluster from our GMM.
Human protein function: Chemokines are chemoattractant cytokines that guide specific immune cells to sites of injury or infection by binding cell surface receptors and triggering intracellular signaling 102–103.
Human protein superfamily: Chemokines are part of the interleukin-8-like chemokine superfamily (SSF54117). The human genome encodes at least 49 proteins in this superfamily 36.
Prediction of viral mimicry: Molluscum contagiosum virus protein MC148R has 25% identity to a chicken CC cytokine 104. It retains the amino acids involved in disulfide bond formation classic to human CC chemokines 104.
Experimental evidence of mimicry: In contrast to human chemokines, the MC148R viral chemokine binds human chemokine receptors typically bound by CC and CXC chemokines (CCR1, CCR2, CCR5, CCR8, CXCR1, CXCR2, CXCR4) 29. It inhibits the chemotaxis of human monocytes, lymphocytes, and neutrophils by antagonizing CC chemokines (MCP-1, MIP-1α, RANTES, I-309) and CXC chemokines (SDF-1, IL-8) 29.
Our results: Querying with MC148R against the human proteome returns five CC chemokines: CCL5, CCL19, CCL20, CCL26, and CCL28. These human chemokines interact with receptors CCR3, CCR5, CCR6, CCR7, CCR10, and CX3CR1 105; the only overlap with the known binding partners of MC148 is CCR5. One would likely hypothesize that MC148R binds CC and CX3C chemokine receptors based on these results. While it does bind CC chemokine receptors, it actually binds CXC rather than CX3C receptors. Still, it's helpful that the method returned multiple query matches, providing some signal that the viral protein generally mimics chemokines instead of a specific chemokine.
GMM output: We've shared an interactive plot with GMM clustering of Foldseek structural comparison results for the viral MC148R protein here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Querying with a viral protease (coronavirus NSP5)
Human PRSS53 aligned with viral protein NSP5.
Predicted PRSS53 is blue, predicted NSP5 is pink. Aligned with the PyMol CE algorithm.
PRSS53 was the hit with the lowest E-value in the best cluster from our GMM.
Human protein function: Proteases are enzymes that catalyze the breakdown of proteins. They play an important role in protein digestion and turnover and act as signal mediators by cleaving proteins into active forms.
Human protein superfamily: NSP5 is part of the trypsin-like serine protease superfamily (SSF50494). The human genome encodes at least 165 proteins in this superfamily 36.
Prediction of viral mimicry: A previous study found that coronavirus NSP5 has structural similarity to over 50 human proteins based on computational comparison of human and viral crystal protein structures 30.
Experimental evidence of mimicry: None.
Our results: We included two NSP5 proteins (conserved coronavirus proteases) in our search. One protein is encoded by human coronavirus HKU1 and the other by SARS-CoV-2. Both NSP5 proteins were in the same Viro3D cluster, so we ran one GMM. Our search returned hits to the human proteases TYSND1, HTRA2, MST1, and PRSS53, albeit with low query-TM scores (mean query TM-score = 0.36).
GMM output: We've shared an interactive plot with GMM clustering of Foldseek structural comparison results for viral NSP5 proteins here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Open question
Do you interpret the relationship between coronavirus NSP5 and human proteases as a potential case of mimicry or generic structural conservation?
Querying with an RNA methylase (coronavirus NSP16)
Human MRM2 aligned with viral protein NSP16.
Predicted MRM2 is blue, predicted NSP16 is pink. Aligned with the PyMol CE algorithm.
MRM2 was the hit with the lowest E-value in the best cluster from our GMM.
Human protein function: RNA methyltransferases catalyze the transfer of a methyl group to RNA molecules to promote RNA regulation.
Human protein superfamily: NSP16 is part of the S-adenosyl-L-methionine-dependent methyltransferases superfamily (SSF53335). The human genome encodes at least 144 proteins in this superfamily 36.
Prediction of viral mimicry: A previous study found that coronavirus NSP16 has structural similarity to over 30 human proteins based on computational comparison of human and viral crystal structures 30.
Experimental evidence of mimicry: None.
Our results: We included two coronavirus NSP16 RNA methylases in our search. One protein is encoded by human coronavirus HKU1 and the other by SARS-CoV-2. Both NSP16 proteins were in the same Viro3D cluster, so we performed one round of modeling. Our search returned hits to the human proteins MRM2, METTL27, CARM1, and TOMT, which all encode methyltransferases. However, these hits had the lowest query TM-score of any returned cluster (mean = 0.31).
GMM output: We've shared an interactive plot with GMM clustering of Foldseek structural comparison results for viral NSP16 proteins here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Open question
Do you interpret the relationship between coronavirus NSP16 and human RNA methylases as a potential case of mimicry or generic structural conservation?
Results for viral proteins with common domains
We explored viral proteins that we didn't expect to be mimics, but that we hypothesized would share some structural similarity with human proteins due to conserved functions across humans and viruses. We had two examples of these proteins: a viral kinase and a viral helicase (key info listed in Table 3). We find that while the kinase had low structural similarity to human proteins, the helicase appears to be very structurally similar to human helicase domains, potentially fitting our definition of mimicry. For both proteins, we show the GMM clustering of matches as well as the most relevant structural alignments of viral to human proteins.
Querying with a viral helicase (pegivirus viral N-terminal helicase domain of the DEAD-box helicase superfamily)
Human DHX9 aligned with a viral helicase.
Predicted DHX9 is blue, predicted helicase is pink. Aligned with the PyMol CE algorithm.
DHX9 was the hit with the lowest E-value in the best cluster from our GMM.
Human protein function: Helicases are enzymes that unwind double-stranded DNA or RNA.
Human protein superfamily: Helicases are part of the P-loop-containing nucleoside triphosphate hydrolases superfamily (SSF52540). The human genome encodes over 1,000 proteins in this superfamily 36.
Prediction of viral mimicry: This isn't a known mimic. We included it because helicases are common to both human and viral proteomes, and we wanted to see how a common domain would perform in our pipeline.
Experimental evidence of mimicry: None.
Our results: We included the pegivirus N-terminal helicase domain of the DEAD-box helicase superfamily in our search. Querying with the viral helicase returned 18 ATP-dependent RNA helicases (DHX proteins, TDRD9, MTREX, YTHDC2). The mean query TM-score for these hits was higher than the mean query TM-score for some mimics with known best matches, such as CD47 (helicase mean = 0.65; CD47 mean = 0.68). This similarity could either reflect viral structural mimicry to human DEAD-box helicases or strong conservation of the structure of the protein to maintain its functional profile.
GMM output: We've shared an interactive plot with GMM clustering of Foldseek structural comparison results for the pegivirus helicase here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Open question
Does the high query TM-score between pegivirus helicase and human helicases indicate a potential case of mimicry?
Querying with a viral kinase (Epstein–Barr virus BGLF4)
Human CDK5 aligned with viral protein BGLF4.
Predicted CDK5 is blue, predicted BGLF4 is pink. Aligned with the PyMol CE algorithm.
CDK5 was the hit with the lowest E-value in the best cluster from our GMM.
Human protein function: Kinases are a conserved superfamily of proteins that catalyze the phosphorylation of specific substrates, mediating signaling or other regulatory processes in cells.
Human protein superfamily: Kinases are part of the protein-kinase-like superfamily (SSF56112). The human genome encodes at least 653 proteins in this superfamily 36.
Prediction of viral mimicry: This isn't a known mimic. We included it because kinases are an enzyme class common to both human and viral proteomes, and we wanted to see how a common domain would perform in our pipeline.
Experimental evidence of mimicry: None.
Our results: Querying with the BGLF4 Epstein–Barr viral kinase returned human CDK5 and non-specific serine/threonine protein kinase (Q59FN2). The mean query TM-score of this match was lower than many well-characterized mimics (kinase mean = 0.36, well-characterized hit mean = 0.64). This likely reflects that while these proteins belong to the same superfamily, they may have different functions.
GMM output: We've shared an interactive plot with GMM clustering of Foldseek structural comparison results for the viral BGLF4 protein here. Each point represents one viral–human protein comparison. Hover over a point to see protein names. Each color represents a cluster from GMM, with the “best” cluster in orange.
Conclusions and next steps
We set out to explore how structural mimicry in parasite proteins might reveal new ways to influence the human immune system. To do this, we developed a computational pipeline to detect mimics and benchmarked our pipeline with a select set of viral proteins.
We found:
- Our method reliably identifies known viral mimics, recapitulating many established relationships in a single analysis.
- There is no clear threshold between true mimicry and generic protein similarity — the user must set their own thresholds based on the goals of their analysis.
We’re icing this work at Arcadia because it doesn’t leverage the unique strengths of our platform, but the pipeline is ready to be used to search for novel mimics across any human-infecting virus. It can also be applied to other parasites, like ticks, though anyone attempting this will need to take care to account for the shared ancestry between all eukaryotes. We think using non-parasites as “negative controls” could be helpful here, but haven’t tried this ourselves.
Be the first to comment on this publication.